
서론
한우의 개량은 인공수정 기법의 발달과 보급에 따라 1985년부터 보증씨수소(Korean Proven Bull; KPN)를 선발 및 활용하는 수소 중심의 개량으로 본격 시작되었다(Park et al., 2013). 초기 한우 개량은 혈통과 표현형 정보만을 이용하여 당대검정과 후대검정으로 이루어진 능력검정체계를 통해 육량 중심으로 이루어졌고, 여러 개선 과정을 거쳐 대용량 유전체정보를 활용하여 추정된 유전체육종가를 이용한 개량이 실시되고 있다(Lee et al., 2014; Chung et al., 2018). 유전체육종가를 추정하여 유전 능력을 평가하는 방법은 혈통 오류에 의해 발생할 수 있는 문제점을 줄이고 혈통육종가에 비해 더 높은 정확도를 보이며(VanRaden, 2008; Choi et al., 2017; Shin et al., 2018a), 세대 간격의 단축으로 인해 유전적 개량량을 보다 더 증가시킬 수 있게 된다(Meuwissen, 2003). 한편으로는 유전능력이 우수한 특정 보증씨수소의 선호 활용으로 한우의 유전적 다양성 감소가 나타날 수 있으니 주의해야 한다는 여러 보고가 있어 왔다(Li and Kim, 2015; Sudrajad et al., 2017; Hong et al., 2017; Lee and Kong, 2021). 그리고 수소 중심의 개량에서 암소의 개량 필요성이 대두되었고 혈통, 표현형 정보 및 후대 도축성적을 기반으로 한우암소 능력검정사업이 시행되어져 오고 있다. 현재 한우 씨수소 선발에 유전체 정보를 이용한 방법이 적용되고 있으며 암소 개량 및 관리를 위한 유전체 정보 활용에 관한 연구 및 시범사업 등이 진행되고 있다(Shin et al., 2018b; Kim et al., 2021).
연관불균형(Linkage Disequilibrium; LD)은 집단 내에서 서로 다른 유전좌위에 위치한 대립유전자들의 비무작위성 관계를 의미한다(Odani et al., 2006). 연관불균형은 유전자의 이동, 선발, 유전적 부동, 돌연변이 및 유전자 재조합 등에 의해 발생한다(Hedrick, 1987). 연관불균형의 정도는 표준화된 D′과 제곱 상관계수(r2)로 추정할 수 있으며, D′은 대립유전자간 연관성을 통해 과거 재조합율을 측정하는 방법이고, r2은 유전좌위에 위치한 대립유전자 빈도간 제곱 상관계수를 측정하는 방법이다(Bohmanova et al., 2010). 유전체 정보를 활용한 연관불균형 추정은 동일한 품종 또는 다른 품종간 유전적 다양성을 파악할 수 있고 유전체 마커를 통해 다양한 분석이 가능하며 마커도움선발(Marker-Assisted Selection; MAS)과 양적형질좌위(Quantitative Trait Loci; QTL) mapping에 중요한 정보를 제공한다(McRae et al., 2002; Qanbari et al., 2010). 연관불균형은 전장유전체관련성분석(Genome-Wide Association Studies; GWAS) 결과를 해석하는데 사용되며(Bohmanova et al., 2010; Lu et al., 2012), 또한, 유전체 선발이나 유전체 예측의 정확도는 연관불균형에 의존적이며 중요한 지표 역할을 한다(Goddard, 2009; Won et al., 2020). 사람에서 전장유전체(whole-genome)를 이용한 연관불균형 연구가 처음으로 보고되었고(Kruglyak, 1999), 더불어 여러 동물들을 비롯해 가축들의 전장유전체를 이용한 연관불균형 추정 연구가 수행되어져 왔다(Kim and Kirkpatrick, 2009; Alvarenga et al., 2018; Muñoz et al., 2019; Jasielczuk et al., 2020).
유효집단크기(Effective population size; Ne)는 많은 세대를 거쳐도 집단 내 유전적 구성이 변하지 않고 유지되는 개체수의 최소 숫자를 의미한다. Ne를 추정함으로써 집단의 진화양상 및 크기 변화 등 집단의 특성 등을 알 수 있고, 복합형질들의 기초가 되는 유전체 구조에 대한 이해와 모델링을 개선하는데 도움이 된다(Tenesa et al., 2007). Ne는 집단 통계적 추정, 혈통 기반 추정 및 유전체 마커 기반 추정 세 가지 방법으로 추정할 수 있다. 집단 통계적 추정과 혈통 기반 추정법은 혈통 완성도에 의존하기 때문에 혈통을 철저하게 관리하고 있는 집단이 아닐 경우 불확실한 혈통으로 인해 유효집단크기 추정 과정에서 오류가 발생하게 된다. 불완전한 혈통 완성도에 의한 오류를 극복하기 위하여 유전체 정보를 기반으로 추정하는 유전체 마커 기반 추정법이 개발되었다(Flury et al., 2010). 유전체 마커 기반 추정법은 유전자 좌위와 대립유전자수의 제곱에 따라 정확도가 증가하고 샘플의 수가 많을수록 정확도가 급격히 증가하게 된다(Waples and Do, 2010). 정확도의 증가를 위한 유전체 정보를 활용하는 방법 중에서도 LD 정보를 기반으로 추정하는 방법이 주로 사용된다.
본 연구는 경북지역 및 전북지역 한우암소 집단의 SNP 정보를 활용하여 한우암소의 유전적 특성을 알아보고자 LD와 Ne를 분석하였다. 한우암소 집단의 SNP정보를 활용하여 분석을 진행한 사례는 아직 미흡한 실정이라고 할 수 있다. 유전체 선발 방법을 통한 유전체 육종가 및 유전체 육종가의 정확도 등은 SNP 마커의 종류, 마커간 간격과 LD 정도, 그리고 개수 등 여러 요인들에 의해 영향을 받으므로 고려해야 할 사항들이 많다고 볼 수 있다. 따라서 농가단위의 한우암소에 대한 유전체 선발의 효율적 적용을 위해 암소 집단의 유전체 구조적 특징 및 유전적 다양성을 규명하고 그에 필요한 기초 자료를 제시하고자 연구를 수행하였다.
재료 및 방법
결과 및 고찰
1. 공시재료 및 SNP 유전자형 결정
경북지역과 전북지역에 소재하는 농가들의 한우암소를 이용하였으며 각각 경북지역 2,076두, 전북지역 941두이었다. 경북지역 한우암소(Gyeongbuk Hanwoo cow; GBH) 2,076두는 sire 336두와 dam 2,076두의 후대축들이고 sire당 후대 암소 약 6.2두, dam당 후대 암소 1두로 이루어져 있다. 전북지역 한우암소(Jeonbuk Hanwoo cow; JBH) 941두는 sire 101두와 dam 779두의 후대축들이고 sire당 후대 암소 약 9.3두, dam당 후대 암소 약 1.2두로 이루어져 있다. 이들의 모근을 채취하여 분석에 필요한 genomic DNA를 추출하였으며, genomic DNA를 추출한 후 경북지역 한우암소(GBH)는 Illumina Hanwoo SNP50K BeadChip (Illumina, San Diego, CA, USA)을 이용했고, 전북지역 한우암소(JBH)는 Illumina Bovine SNP50K Beadchip (Illumina, San Diego, CA, USA)를 이용하여 SNP 유전자형을 결정하였다. Hanwoo SNP50K chip과 Bovine SNP50K chip간의 공통된 SNP 마커의 수는 48,642개이었으며, 이후의 분석을 위해 Genome Studio 2.0 (Illumina, San Diego, CA, USA) 프로그램을 이용하여 final report를 생성하였다.
2. SNP 품질관리 (Quality Control; QC)
분석에 유용한 SNP을 선별하기 위해 Plink v1.9 (Purcell et al., 2007)를 이용하여 SNP 및 샘플의 품질평가를 실시하였다. Call rates <0.9, Minor Allele Frequency (MAF) <0.05, Hardy-Weinberg Equilibrium (HWE) p-value<1 × 10 − 6, 성염색체, 미토콘드리아 염색체 및 식별 불가능한 데이터들을 사전에 제외하였다.
3. 연관불균형 추정
연관불균형의 정도는 Plink v1.9를 이용하여 r2 (Hill and Robertson, 1968) 방법으로 분석하였다. r2을 이용하기 위해 D는 다음과 같은 모델을 이용하였다(McVean, 2008).
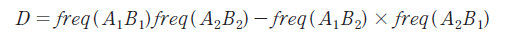 식 (1)
식 (1)
여기서, freq(A1B1)과 freq(A2B2), freq(A1B2)과 freq(A2B1)는 집단내 A와 B 두 유전자 좌위에 해당하는 각 대립유전자로 이루어진 일배체형의 빈도 그리고 r2을 이용하여 서로 다른 두 유전자 좌위 A와 B의 연관불균형의 정도를 계산하였다(Hill and Robertson, 1968).
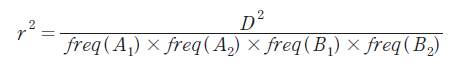 식 (2)
식 (2)
여기서, freq(A1), freq(A2), freq(B1), freq(B2)은 집단 내에서 A1, A2, B1, B2 대립유전자의 빈도수
4. 유효집단의 크기 추정
유효집단의 크기(Ne)는 SNeP v1.1 (Barbato et al., 2015)를 통해 SNP간의 연관불균형 정도와 재조합율을 기반으로 과거 세대로부터 최근 세대까지 지남에 따라 과거 유효집단의 크기를 추정하는 방법을 이용하였다(Corbin et al., 2012). Ne는 분석프로그램의 마커간 최대거리 설정 값이 4,000 Kb이므로 과거 13세대부터 분석하였고 모두 기본값으로 설정하였다.
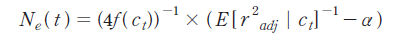 식 (3)
식 (3)
여기서, Ne(t)는 t세대 이전에 추정된 유효집단의 크기
ct: 마커간의 물리적 거리로 정의된 재조합 비율
r2adj: 샘플 크기에 맞게 보정된 연관불균형
α: 돌연변이 발생에 대한 보정 값, 1로 설정
1. SNP 유전자형 결정
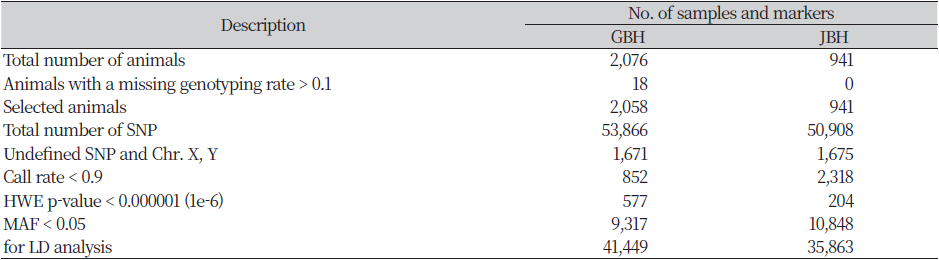
한우암소 집단의 상염색체, 성염색체 및 미토콘드리아 염색체 등을 모두 포함하여 SNP 유전자형을 결정했고 경북지역 한우암소(GBH)는 총 53,866개의 SNP, 전북지역 한우암소(JBH)는 총 50,908개의 SNP가 식별되었다. QC 과정을 진행한 결과 총 유전자형 결정율은 GBH 0.9917, JBH 0.9849였다. GBH는 2,076두 중 18두가 유전자형 결정율 기준치에 미달되어 2,058두가 선별되었고, 12,417개의 SNP이 제외되었으며 41,449개(전체 SNP 중 76.95%)의 SNP을 분석에 이용하였다. JBH는 941두 모두 유전자형 결정율 기준치에 부합하였고, 15,045개의 SNP이 제외되었으며 35,863개(전체 SNP 중 70.45%)의 SNP을 분석에 이용하였고, GBH 및 JBH 집단에서 공통적으로 사용된 SNP 마커의 개수는 34,813개이었다(Table 1). QC 기준을 GBH와 JBH 모두 동일하게 설정하였으나, 분석에 이용된 지역간 SNP 개수의 차이는 SNP chip의 종류(Hanwoo SNP chip/Bovine SNP chip)에 주로 기인한 것으로 사료된다.
2. 상염색체에 위치하는 SNP 정보
각 상염색체별(n=29) SNP의 개수, 총 길이, MAF, 평균 간격 정보를 요약했다(Table 2). 상염색체내 SNP 평균 개수는 1,429개(GBH), 1,237개(JBH), SNP간의 거리차이로 구한 염색체의 총 길이는 2508.56 Mb (GBH), 2507.35 Mb (JBH), 평균 MAF는 GBH와 JBH가 모두 약 0.27 (0.268/0.271)로 나타났고, SNP간 평균 간격은 0.061 ± 0.016 Mb (GBH), 0.070 ± 0.084 Mb (JBH)로 측정되었다. 두 지역 한우 모두 1번 염색체의 SNP 개수가 2,600개(GBH), 2,406개(JBH)로 가장 많았고, GBH는 28번 염색체의 SNP 개수가 720개, JBH는 25번 염색체의 SNP 개수가 665개로 각각 가장 적게 나타났다. 염색체 길이는 두 지역 모두 1번 염색체가 158.23 Mb로 가장 길었으며, 25번 염색체가 42.70 Mb (GBH), 42.36 Mb (JBH)로 각각 가장 짧게 나타났다. MAF 평균값은 GBH가 7, 21, 23, 25번 염색체에서 약 0.28로 가장 높았고 14번 염색체에서 0.249로 가장 낮았으며, JBH가 25번 염색체에서 약 0.29로 가장 높았고 9번 염색체에서 0.260으로 가장 낮았다. 경북 및 전북의 두 지역 간의 차이는 사용한 SNP chip의 종류, 즉 chip에 탑재된 SNP 마커의 개수와 특성에 기인한 것으로 사료되며, 국내 한우를 대상으로 연구한 Sudrajad et al. (2017), Hong et al. (2017), Lee and Kong (2021)의 결과와 비교해 보면 모두 이번 연구결과와 아주 유사하였고, 또한 Bos taurus (cattle) ARS-UCD 1.3 (NCBI, 2018) 및 Bos_taurus_UMD_3.1.1 (NCBI, 2014)의 보고와 크게 차이 나지 않는 양상을 나타냈다. 외국 소 품종을 이용한 Saravanan et al., (2022), Singh et al., (2021) 및 Jemma et al., (2019) 등이 보고한 결과와 아주 유사하였으나, Salem et al., (2018) 및 Jasielczuk et al., (2016)의 보고와는 다른 양상인데, 특히 Bos taurus 품종에 속하는 앵거스, 샤롤레 및 이들의 교잡종의 비교적 많은 두수(약 1,600두 이상)를 이용하여 보고한 Lu et al. (2012)과 비교해 보면, 다른 부분은 아주 유사한 결과를 보였으나 염색체의 총길이가 2,534.98–2,535.30 Mb로 이번 연구결과보다 약간 크게 분석되었다. 이는 SNP간 평균 간격이 약 70 kb로 나타나 이번 연구(평균 약 65 kb) 보다 SNP 간격이 넓어진 것과 분석 품종 및 chip에 탑재된 SNP 마커 종류 등의 차이인 것으로 사료된다.
|
Table 2. Summary of the SNP markers analyzed for each autosomal chromosome 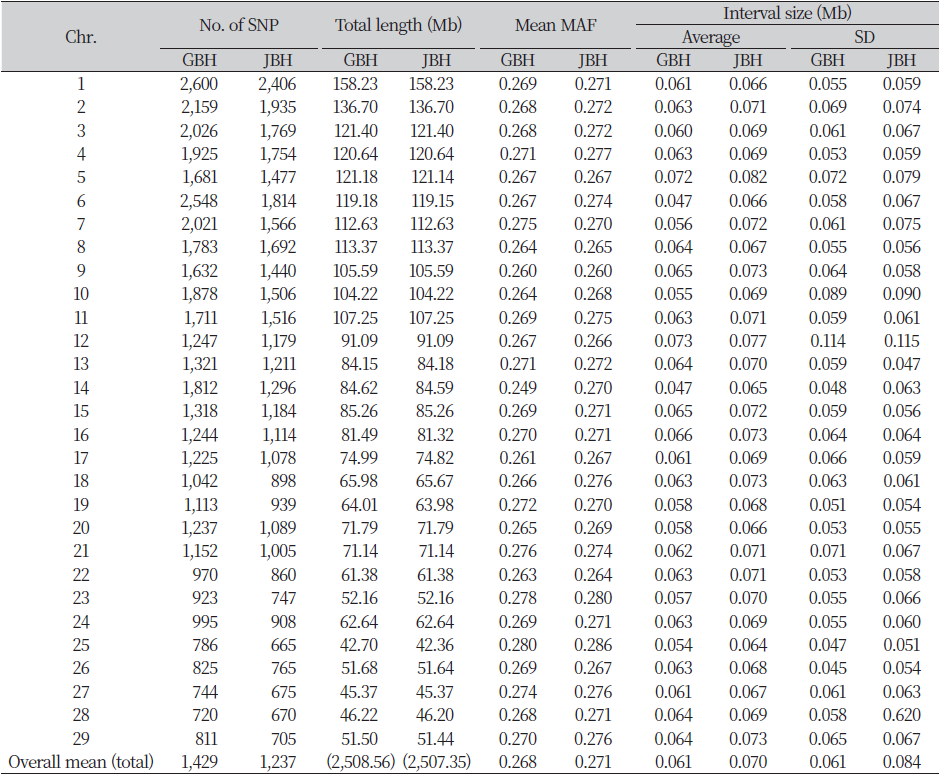
|
|
GBH: Gyeongbuk Hanwoo, JBH: Jeonbuk Hanwoo, Chr.: Chromosome, MAF: Minor Allele Frequency |
3. 대립유전자 빈도 분석
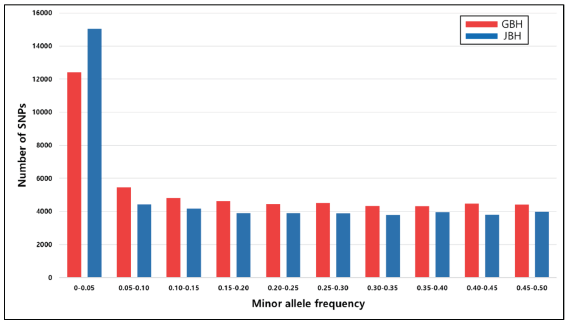
MAF를 0-0.5까지 10개의 범위로 구분하여 SNP의 분포를 나타냈다(Figure. 1). 두 지역 한우 모두 0-0.05 범위에서 가장 많은 SNP이 확인되었고 이를 제외한 모든 범위에서 MAF는 고른 분포를 보였다. 그리고 0-0.05 범위에서 GBH가 JBH보다 적은 SNP 개수를 보였으나 나머지 범위에서 더 많은 SNP 개수를 나타냈다. MAF 0.05 이후 범위에서 이러한 양상을 보인 것은 SNP chip에 위치한 마커간의 거리가 일정하게 위치해 있으므로 나타난 결과로 사료된다. Kim and Kirkpatrick (2009)은 MAF 0-0.05 범위에서 SNP의 분포가 가장 낮게 나타났고, 0-0.05 범위 이후로 SNP의 분포가 높게 나타났다. Cho et al. (2012)은 MAF 0-0.05 범위에서 SNP의 가장 낮은 분포를 보였고, 나머지 범위에서는 고른 분포를 보였다. 반대로 Li and Kim (2015), Hong et al. (2017), Lee and Kong (2021)은 MAF 0-0.05 범위에서 SNP 개수가 가장 높은 분포를 보였고, 나머지 범위에서 고른 분포를 나타내 이번 연구결과와 비슷한 경향을 보였다. Espigolan et al. (2013)은 MAF 0.05-0.10 범위에서 가장 높은 SNP의 분포를 보이다가 0.10 범위 이후로 낮은 SNP의 분포를 나타냈다. Saravanan et al. (2022)는 MAF 0-0.05 범위에서 가장 높은 SNP의 분포를 보였으나 나머지 범위에서 비슷한 분포를 나타냈다. 이와 같이 한우 품종간 결과와 타품종간 결과의 차이는 사용된 분석 두수, SNP chip의 종류(제조회사 및 SNP의 개수) 및 각 품종별 적합도에 따른 차이점으로 사료된다. 또한, Scheet and Stephens (2006)는 MAF가 유전자 마커의 변이성을 간접적으로 내포하고 있으며 MAF가 낮을 경우 연관불균형의 정도를 낮출 수도 있다고 보고했다.
4. 연관불균형 추정
SNP쌍 사이의 거리를 50 kb 기준으로 하여 추정한 정보를 표로 나타냈다(Table 3). 총 SNP쌍 개수는 각각 776,966개(GBH), 530,350개(JBH)로 식별되었고 평균 r2이 GBH가 0.05 ± 0.12, JBH가 0.04 ± 0.09로 나타났다. 50-100 kb 거리에서 SNP쌍의 개수는 각각 45,083개(GBH), 28,176개(JBH)로 가장 많았고, 950-1,000 kb 거리에서 각각 36,596개(GBH), 26,113개(JBH)로 가장 적게 나타났다. 0-50 kb 거리에서 r2이 각각 0.230(GBH), 0.206(JBH)으로 가장 높았고, 950-1000 kb 거리에서 각각 0.028(GBH), 0.019(JBH)로 가장 낮게 나타났다. SNP쌍 개수와 r2 모두 SNP간 거리가 멀어짐에 따라 감소하는 추세를 보였다. 평균 r2이 GBH는 0-50 kb 거리에서 0.230이었으나 50-100 kb 거리에서 0.119로 급격히 감소했고, JBH가 0-50 kb 거리에서 0.206이었으나 50-100 kb 거리에서 0.114로 급격하게 감소함에 따라 LD가 붕괴됨을 알 수 있었다. 대체적으로 GBH가 JBH보다 높은 값을 나타냈으며 이는 이용한 SNP chip 종류와 SNP 마커의 차이로 사료된다. 한우 거세우를 이용해 LD를 분석한 연구들은 SNP간의 거리에 따른 LD가 각각 0.1-0.2 Mb (Cho et al. 2012), 40-60 kb (Li and Kim, 2015), 50-100 kb (Hong et al. 2017; Lee and Kong, 2021) 거리에서 급격하게 감소하는 결과를 나타냈으며 Hong et al. (2017)과 Lee and Kong (2021)이 이번 연구결과와 유사하게 붕괴되는 결과를 나타냈다. 타품종 결과를 살펴보면, 유럽종에 속하는 앵거스, 샤롤레 및 이들의 교잡종을 분석한 Lu et al. (2012)의 보고와도 유사한 결과를 보이고 있다. Vrindavani 교잡종을 연구한 Singh et al. (2021)는 10-25 kb 거리에서 평균 r2이 급격하게 줄어드는 결과를 보였다. Qanbari et al. (2010) 는 모든 SNP쌍 기준에서 SNP쌍 사이의 거리가 멀어질수록 SNP쌍 개수는 늘어나는 결과를 보였고 평균 r2은 서서히 감소하는 경향을 보였으며 LD 패턴을 분석함으로써 적절한 연관성 분석을 위한 r2은 0.25 이상 되어야 한다고 보고했다. 또한, Meuwissen et al. (2001)은 r2이 0.2 이상이어야 유전체 육종가 정확도가 85%에 도달할 수 있다고 보고했고, 최근 여러 소 품종의 전장유전체 분석 연구들은 다양한 고해상도 SNP chip을 사용하는 경향을 나타내고 있으므로(Xu et al., 2019; Mokhber et al., 2019) 한우의 경우에도 추후 더욱 유의한 전장유전체 연관성 분석과 유전체 선발을 위해 고해상도이며 한우 맞춤형 버전의 SNP chip이 imputation 등 필요할 것으로 사료된다.
|
Table 3. Pairwise linkage disequilibrium (r^2) for SNP at every 50 kb distance 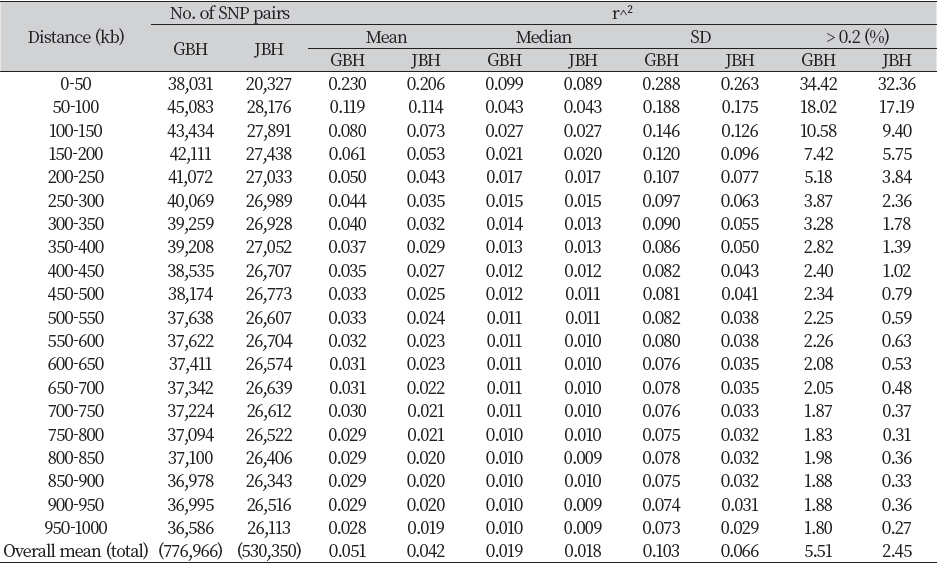
|
|
GBH: Gyeongbuk Hanwoo, JBH: Jeonbuk Hanwoo, SD: Standard deviation |
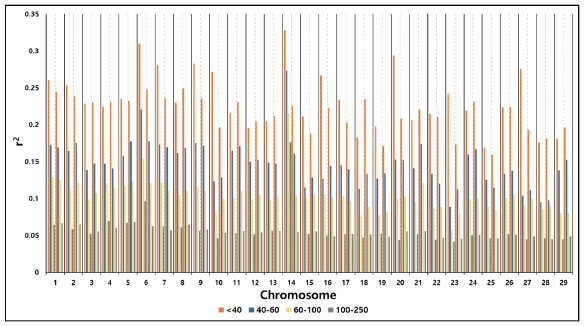
염색체에 따른 LD 분석 결과 정보를 40 kb 미만, 40-60 kb, 60-100 kb, 100-250 kb 기준으로 요약했다(Fig. 2). 그 중 40 kb 미만에서 r2은 GBH가 0.170-0.328까지 나타났고 JBH가 0.160-0.249까지로 나타났다. r2이 0.25 이상으로 나타난 염색체는 GBH가 1, 2, 6, 7, 9, 10, 14, 16, 20, 27번 염색체로 총 10개, JBH에서는 검출되지 않았다. 또한, r2이 0.2 미만인 염색체는 GBH가 12, 18, 19, 25, 28, 29번 염색체가 나타났고, JBH가 10, 15, 19, 23, 25, 17, 28, 29번 염색체가 나타났다. Li and Kim (2015)은 r2이 0.25 이상인 염색체가 나타나지 않았고, Hong et al. (2017)은 r2이 0.25 이상인 염색체가 6번 염색체(0.254)로 식별되었다. 본 연구에서 GBH의 경우 r2이 0.3 이상인 염색체가 6번, 14번 염색체로 나타났는데, 이들 염색체는 소의 도체중 형질과 연관성 있는 SNP이 위치한 것으로 보고되었으며(Edea et al., 2018), 추후 GWAS 연구에 적용할 수 있을 것으로 사료된다. 그리고 GBH와 JBH가 r2 기준에 따라 염색체의 개수가 다른 양상을 보였는데 이는 분석에 이용한 SNP chip의 품종 특이성 및 SNP 밀도(density of SNP), 분석 두수와 밀접한 관련성이 있는 것으로 판단된다.
5. 유효집단의 크기 추정
연관불균형의 정도를 바탕으로 유효집단의 크기를 추정하여 세대별 유효집단의 크기를 나타냈다(Fig. 3). 추정 결과 GBH가 69세대 전(69 generations ago) Ne는 994두였으나 13세대 전 Ne는 267두로 감소하였고, JBH가 70세대 전 Ne는 1,588두였으나 13세대 전 Ne는 415두로 감소했음을 나타냈다. 두 지역 한우 모두 현세대로 가까워질수록 유효집단의 크기가 급격하게 감소하였다. 13세대 전 Ne가 GBH (267두) 보다 JBH (415두)에서 더 높게 나타났는데, 이는 사용한 SNP chip에 따른 LD (r2)의 차이에서 주로 기인되며 다른 원인으로는 시료수의 차이(2,058두 및 941두)로 사료되어진다. 그리고 지역별 암소집단의 유전적 다양성의 감소 및 근친정도의 차이도 고려할 수 있겠으나 이는 추가연구가 필요하리라 판단된다. 한우 거세우의 Ne를 추정한 연구 결과들을 보면, Lee et al. (2011)는 500세대 전 Ne가 2,743두였으나 3세대 전 Ne가 98두로 감소했다. Cho et al. (2012)는 45~50세대 전 Ne가 1,130두였으나 1~5세대 전 Ne는 84두로 감소했다. Li and Kim (2015)은 3,179세대 전 Ne가 3,802두였지만 4세대 전 Ne는 327두로 나타났다. Sudrajad et al. (2017)는 Brown Hanwoo가 3,694세대 전 Ne가 5,459두였으나 11세대 전 Ne가 531두로 감소하였고, Brindle Hanwoo가 4,756세대 전 Ne가 4,103두였으나 11세대 전 Ne가 53두로 감소하였으며 Jeju Black이 4,669세대 전 Ne가 4,672두였으나 11세대 전 Ne가 60두로 감소한 결과를 나타냈다. 이전 연구 결과들 모두 과거 세대로부터 최근 세대까지 세대가 지남에 따라 Ne가 급격하게 감소하고 있는 양상을 보였다. 타품종의 연구 결과들을 보면, Cañas-Álvarez et al. (2016)는 과거 1000세대부터 200세대 이전까지 Ne가 완만하게 감소하였으나, 200세대 전 이후부터 최근 세대까지 급격한 감소를 나타냈다. Jemaa et al. (2019)는 64세대 전 Ne가 1,410두였으나 최근 1세대 전 Ne가 51두로 감소했음을 보였다. Fabbri et al. (2020)는 이탈리아 토착 4품종 모두 세대가 지남에 따라 Ne의 감소를 보고하였고 그 중에서 Limousin이 가장 급격한 저하를 보였다. 유효집단의 크기는 집단내 동시대에 살아있는 개체 수, 세대 간격 및 집단 크기의 분산에 영향을 받기 때문에 같은 품종 이어도 각 연구결과마다 다른 결과값이 나타나게 된다(Lu et al., 2012). 한우암소는 한우의 선발 과정에 있어 인공수정 기법의 발달로 인해 이러한 결과가 나타났음으로 사료된다(Wang, 2005). 또한, 한우의 유전적 다양성을 유지하기 위해선 세대당 Ne가 최소 50은 되어야 하므로(FAO, 2000) 농가 단위에서 선호하는 보증씨수소 정액만을 이용하여 나타나는 쏠림 현상과 이후 후대 생산에 대한 제도적 장치를 마련하여야 할 것으로 사료된다.
본 연구는 아직까지 한우암소 집단의 유전체 정보를 활용한 기초 연구가 부족하고 유전체 선발을 통한 농가단위 개량이 본격화되기 전 현시점에서 한우암소의 유전적 구조 및 다양성을 규명하기 위하여 연관불균형의 정도와 과거 유효집단의 크기를 분석하였다. 이번 분석 결과 한우암소의 연관불균형이 붕괴되고 유효집단의 크기가 급격하게 줄어들고 있음에 따라 유전적 다양성이 감소하고 있는 상태로 나타났고 과거 한우의 연구 결과와 유사하게 감소하고 있음을 알 수 있다(Lee et al., 2011; Cho et al., 2012; Li and Kim, 2015; Sudrajad et al., 2017; Hong et al., 2017; Lee and Kong, 2021). 타품종의 분석 결과와 비교하면 한우는 연관불균형의 정도가 낮고 최근 세대의 유효집단의 크기가 비교적 크게 나타났는데 이는 타품종에 비해 선발강도가 낮기 때문에 나타난 것으로 판단된다. SNP마커의 종류와 개수, 연관불균형의 정도, 유효집단의 크기 등이 결과적으로 QTL 탐색이나 GWAS 결과 및 유전체 육종가 추정에 영향을 미치기 때문에 본 연구결과를 바탕으로 현재 상용화된 SNP마커의 종류를 변경하여 SNP chip 성능을 향상시키는 방안을 고려해야 할 것이며, 경북과 전북 이외 지역의 한우암소를 통해 유전체 정보를 이용한 inbreeding coefficient, haplotype block structure, runs of homozygosity 등 다양한 연구가 필요할 것으로 사료된다. 그리고 과거 한우를 이용해 연관불균형의 정도와 유효집단의 크기를 분석한 결과들 모두 한우암소에 대한 검정을 필요로 하고 있으므로 더 다양한 집단에 대한 많은 연구를 통해 한우의 유전적 다양성과 근친도를 파악하고 한우암소 맞춤 교배 계획 등의 한우 암소 축군에 대한 지속적인 유지관리가 필요한 것으로 사료된다.

