서론
최근 유전체정보를 이용한 선발 방법인 유전체 선발 (Genomic Selection)을 적용하기 위해 많은 노력들을 하고 있다. 유전체육종가를 추정하기 위한 방법으로는 유전체 정보만을 이용하여 GRM를 구성하는 GBLUP(Genomic BLUP) 방법과 추정된 육종가(Traditional EBV)와 유전체 정보를 이용하여 추정된 MBV(Molecular Breeding Value)를 혼합 공식 (Blending Formula)을 이용하여 계산하는 Two-step 방법과 기존의 혈연관계 (NRM) 매트릭스를 대신하여 NRM과 GRM (Genomic Relationship Matrix)을 혼합하여 만들어진 H-matrix를 이용하는 single-step GBLUP (Misztal et al., 2009)이 있으며, 최근에는 혈연관계를 이용하여 유전체 정보가 없는 개체의 유전체 정보를 생성하여 계산하는 single-step Super Hybrid 모델 (Fernando et al., 2014)을 이용하여 유전체육종가를 추정하고 있다. 유전체 정보를 활용하는 경우 기존 육종가에 비해 더 높은 신뢰도를 가지며 출생 시 조기 선발에 이용하여도 편의가 발생하지 않는 장점이 있다. 현재 우리나라에서 한우의 개량은 자손의 성적을 이용하여 개체의 능력을 간접 예측하는 후대검정시스템을 적용하고 있으며, 후보KPN의 나이가 약 5년 되었을 때, KPN 당 10두 내외의 후대검정우에 대한 표현형 성적을 얻게 되며 약 70%의 신뢰도로 개체의 유전평가가 실시되고 있다. 한우의 세대 당 유전적 개량량을 향상시키기 위해서는 유전평가의 신뢰도를 향상, 선발강도 향상, 세대간격 단축 등의 노력이 필요하다. 가축에서 유전체 선발의 적용은 기존의 BLUP 추정방법에 비하여 신뢰도 향상 및 세대기간 단축을 통해 높은 유전적 개량량을 얻을 수 있으며(Schaeffer, 2006; Hayes et al., 2009), 특히 유전력이 낮은 형질, 개체의 생을 마감한 후에 생성되는 도체형질, 표현형 자료수집에 위험이 따르는 질병저항형질에 대해 더욱 유용하게 활용될 수 있다(Meuwissen et al, 1996; Dekkers et al, 2002). 현재까지 혈통 및 표현형자료를 이용하여 EBV(Estimated Breeding Value)를 추정하여 개체의 선발과 도태를 실시하였지만, 최근 유전체 자료를 기반으로 Single Step Method를 사용하여 표현형, 혈통 및 유전체 마커의 정보를 기반으로 하는 유전체 평가를 많이 사용하고 있다. Single Step Method는 전통적인 방식인 혈통 기반에 유전체 마커 정보를 추가하여 평가할 수 있으며, 이는 혈연관계행렬(Relationship matrix)과 호환되기 위해 혈연관계행렬을 조정하여 평가 방법을 제시하고 있다. 이 방법은 유전자형 자료의 단변량 및 다변량 모델에 대하여 모두 비교가 가능하다. 이를 통해 마커 정보가 있는 Single Step Method는 혈통 기반 방법에 비해 예측 능력을 향상 시킬 수 있으며, 혈통을 가진 모든 개체에 대해 향상된 예측값을 제공받을 수 있다(Christensen et al, 2012).
ssGBLUP은 유전체 평가에 선호되는 방법(Aguilar et al., 2010; Christensen et al. 2010)이며, 이는 유전체육종가를 추정 시 유전자 마커 및 혈통 정보를 동시에 포함하여 평가하고 혼합모형식을 풀기 위한 방법으로 유전체와 관련된 G matrix를 역행렬하여 추정한다. 역행렬 계산과 혼합모형식의 반복적 풀이로 G-1 행렬로 인한 고정된 연산의 수는 유전자형을 가진 개체 수에 따라 증가하게 된다. 일반적으로 유전자형을 가진 개체 수가 30,000개를 초과하면 혼합모형식 분석 시간이 G-1 행렬에 의해 좌우되기 때문이다. 이러한 문제를 완화하기 위해 Legarra et al.(2012)는 몇 가지의 Single Step 혼합모형식을 제시하였으며, 그 중 일부는 G-1 행렬을 없애거나 무효화시키는 방법도 있다(Liu et al., 2014; Fernando et al., 2014).
Misztal et al.(2014) 및 Fragomeni et al.(2015)는 대규모 유전자형 개체군에 대한 추정을 가능하게 하는 검증된 최신 알고리즘(APY)을 발표하였으며, 이는 행렬의 근사된 희소행렬의 역행렬을 사용하도록 제안하였다(Masuda et al., 2016; Strandén et al., 2017). PCG 방법에 의한 반복적인 풀이과정에서 Single Step MME는 일반적인 최소한의 반복 회수를 제공하게 되며(Legarra and Ducrocq, 2012; Strandén and Mäntysaari, 2014), 이는 PCG에서 원래 MME와 동일한 수렴 특징을 가지지만 계산상 간단한 ssGTBLUP이라는 정확한 접근법을 제시하고 있다. 유전체 평가에서 있어 일반적인 평가는 유전체 혈연관계행렬을 가진 BLUP 또는 SNP BLUP 모형을 기초로 한다. Multi-Step 유전체 평가를 하기 위해, 상기 언급한 두 가지의 유전체 모형은 유전체 참조집단에 대해서도 동일한 예측치를 제공하는 것이 입증되었고, 두 가지 모형의 동일한 결과치는 표현형이 없는 어린 유전체 개체에서도 검증이 되었다. 양적형질의 유전 전달을 담당하는 유전자는 SNP 마커의 불완전한 연관불평형과 돌연변이로 인한 SNP 마커들의 유전분산으로 모두 설명할 수는 없다. RPG effect는 일반적으로 불완전한 연관불평형을 설명하기 위한 유전체 모형에 적합하다. 신뢰도 추정에 있어 Multi-Step GBLUP 및 SNP BLUP 모형이 RPG effect를 포함하는 경우 유전체 참조집단과 동일한 예측을 나타낸다는 전제로 이 두 가지 유전체 모형 모두 다양한 유전자를 가지고 있는 형질의 동일성 효과가 표현형이 없는 어린 개체에 대해서도 동일하게 검증이 되었다(Liu, 2017).
본 연구에서는 한우농가에서 직접 사육된 한우의 유전체 자료, 도축성적 및 혈통자료를 수집하여 Single Step 방법을 이용하여 한우의 유전체육종가를 추정하고 RPG effect를 이용하여 신뢰도를 추정하여 농가에서 직접적으로 활용 가능한 유전체 유전능력 추정을 위한 기초자료로 제공하고자 본 연구를 실시하였다.
재료 및 방법
1. 공시자료
본 연구를 위하여 전국의 한우농가에서 다양한 방법으로 채취된 8,413두의 DNA를 추출하였다. 유전체 정보를 생성하기 위해 Axiom Bovine 60k 버전3(Affymetrix Inc., 2006) SNP패널을 이용하였다. 유전체 분석의 정확성을 높이기 위한 Quality Control은 SNP가 성염색체에 상에 있거나 염색체상의 위치(Position)가 확인되지 않은 SNP를 제거하였으며, SNP Call rate가 95% 이하, Minor Allele Frequency (MAF)가 0.01 이하, Hardy-Weinberg 불평형(HWE)을 측정한 Chi-square (χ2) 값이 95%를 초과한 마커를 제거한 총 64,973개의 SNP 마커를 이용하였다. 유전체 정보를 가진 8,413두 중 중복개체, 혈통오류, 종속변수와 매칭이 되지 않는 개체는 제거하여 유전체 정보가 있는 참조 축군 6,616두를 분석에 이용하였다. 분석에 사용된 보증KPN는 정액을 이용하여 DNA추출을 진행하였으며, 거세우는 축산물품질평가원에서 제공받은 조직 샘플을 이용하여 DNA추출을 실시하였고, 암소의 경우, 모근을 채취하여 모근에 있는 DNA를 추출하여 분석에 이용하였다.
혈통자료의 경우 6,616두와 관련된 5계대의 자료를 한국종축개량협회에서 수집하였다. 분석에 이용된 자료는 총 5,153,168두에 대한 혈통자료를 이용하였다. 도체자료는 5,153,168두 중 도체자료를 가지고 있는 개체 중 결측치 및 이상치를 사전제거 한 후 남은 2,376,865두에 대해서 축산물품질평가원에서 자료를 수집하여 분석에 이용하였다. 이용된 형질은 도체중(Carcass Weight, CW), 등심단면적(Eye Muscle Area, EMA), 등지방두께(Backfat Thickness, BF) 및 근내지방도(Marbling Score, MS)의 4개 형질을 고려하였다.
2) 분석방법
① ssGBLUP
ssGBLUP을 이용한 평가 모델은 다음과 같다.

여기서, y는 표현형 자료 벡터, b와 u는 각각 고정효과와 임의효과에 대한 추정치 벡터, X와 Z는 각각 b와 g에 대한 계수 행렬, e는 잔차에 대한 벡터이다. 또한, GRM를 구성을 위해 Van Raden (2008)이 제안한 방법은 다음과 같다.

여기서, pi는 i번째 SNP 마커의 대립유전자 빈도(allele frequency), Z는 마커에 대한 근접행렬(incidence matrix)이며, 혼합방정식(MME)은 다음과 같다.

하지만, GBLUP의 평가는 유전체 정보가 있는 개체만 모델에 포함되어 평가되는 단점이 있다. 이를 보안하기 위해 Misztal et al (2009)은 GRM과 NRM을 이용하여 유전체 정보가 있는 개체와 유전체 정보가 없는 개체를 동시에 이용하여 육종가를 한번에 추정하는 single-step GBLUP (ssGBLUP)을 제시하였다. ssGBLUP은 BLUP의 NRM과 GRM 대신 Harmonic Matrix를 이용하는 방식이며, 평가에 이용되어지는 H-matrix 역행렬 (H − 1) 추정하는 방법은 다음과 같다.

평가를 하는 개체 수의 증가는 H − 1의 계산 시, 컴퓨터의 성능적인 계산에 어려움이 있지만, Legarra et al.(2012)은 효율적인 H − 1를 만들기 위한 방법을 제안하면서 개체 수가 늘어남에 따라 행렬크기의 증가 문제를 해결할 수 있다. MEM 방법에는 RR-BLUP, SNP-BLUP 및 Bayes 방법이 있다. Bayes 방법은 사전 분포에 적합한 샘플(Random sample)을 추출하여 유전체 정보가 있는 개체보다 SNP 마커의 수가 많은 것과 관계없이 적합한 SNP 효과를 추정하는 것이 가능하다. 상기 모형을 이용하여 GEBV를 추정하는데 MiX99(Lidauer, 2015) 프로그램을 이용하여 추정하였다.
② Reliability with RPG effect
다음의 Mixed linear model(혼합 선형 모형)은 유전체 참조집단의 표현형 데이터를 분석하는데 이용된다(Liu et al., 2014).

여기서
y는 후대성적이 있는 KPN의 역회귀육종가, 정확한 표현형 자료들의 nT× 1번째 벡터(Jairath et al., 1998);
· nT는 표현형 자료들의 수; b는 모든 fixed effects의 nF × 1번째 벡터;
· nF는 모든 fixed effects의 수; μ2는 additive genetic effects의 n× 1번째 벡터;
· n는 reference animals(참조집단)의 수; e는 residual effects의 nT × 1의 벡터;
·X는 nT × nF 번째 값,
·W2는 nT × n의 b와 μ2 효과들의 incidence matrices 값
상기 유전체 모형은 다형질 유전체 평가로 확장할 수 있다. 표현형 자료가 있는 참조집단의 개체 중에서 몇 가지 형질에 대해 결측치가 있어도 참조모집단에 포함시켜서 평가할 수 있다. 다형질 참조 모집단을 한 번에 평가하는 것이 각 형질별로 분리하여 평가하는 것보다 컴퓨터 연산방법이 효율적이다. 이는 모형에 포함되는 효과들을 병렬구조로 평가할 수 있기 때문에 가능하다. 유전체 평가를 위해 선별된 m SNP 마커는 불완전한 연관불평형 때문에 모든 상가적 유전분산을 설명할 수 없다고 가정한다. 여기서 k는 모든 m SNP 마커에 의해 설명되지 않는 상가적 유전분산의 비율이다. 따라서, 상가적 유전효과는 다음과 같이 나눌 수 있다.

여기서
g는 m SNP 마커의 상가적 유전효과의 m × 1번째 벡터
a2는 참조집단의 RPG 효과의 n × 1번째 벡터
Z는 참조집단의 회귀계수가 포함된 m SNP 마커의 n × m번째 유전자형 design matrix: 2 − 2pj, 1 − 2pj, − 2pj, j번째 마커의 AA, AB, BB의 유전자타입(VanRaden, 2008)
여기서
pj는 j번째 SNP 마커의 allele 빈도이며 DGV를 다음과 같이 쓸 수 있다.

모형식의 u2를 방정식으로 대체하면 SNP BLUP 모형이 된다.

var(e) = R로 두면, 참조집단의 RPG 효과는 정규분포를 갖는다.
a2 ∼ N(0, kσu2A22)여기서, A22는 참조집단 개체들의 혈연계수행렬을 나타낸다.
잔차 유전체 분산성분인(Residual polygenic variance parameter) k는 0과 1 사이의 값을 취할 수 있지만 2개의 경계 값인 k = 0, k = 1은 제외한다. 또한 SNP 마커의 상가적 유전효과는 정규분포를 따른다고 가정한다.

모든 m SNP 마커가 SNP BLUP 모형과 같은 상가적 유전분산을 설명하는 경우, 연관성이 없는 SNP효과의 가정하에서 B는 m × m의 대각선 행렬이다(Meuwissen et al., 2001; VanRaden, 2008; Liu, 2011). 그러면 다음과 같이 나타낼 수 있다.

g와 a2 사이의 공분산을 0이라고 가정할 때:

관측된 유전체 관계행렬은 다음과 같이 표시한다.

Liu et al.(2014)에서 보고한 바와 같이, 다음과 같이 나타낼 수 있다.

상기 공식에서 유전체 관계행렬 G22을 볼 수 있는데 이는 VanRaden(2008)이 처음으로 제시하였고, SNP BLUP 모델에서 가중치 (1-k)를 가지는 유전체 혈연계수행렬과 가중치 k를 가지는 추정 혈통계수행렬 A22의 선형함수이다.
RPG 효과 a2와 상가적 유전효과 u2사이의 공분산은 다음과 같다.

상기 식의 공분산 식을 이용하여, RPG 효과 a2는 GEBV û2에서 예측할 수 있다.

상기 식을 기반으로 계산을 하면, 다음의 식을 얻을 수 있다.

유사한 방법으로 DGV와 u2의 공분산을 얻을 수 있다.

ûDGV 참조집단의 DGV는 û2에서 다음과 같이 예측할 수 있다.

(GDGV) − 1 Z′B에 의한 사전 다중 방정식은 SNP효과 g에 대한 방정식으로 이어진다.

상기 방정식은 VanRaden (2008)이 최초로 제시하였고, 동등한 예측치를 나타내는 공식은 Stranden and Garrick (2009)이 제시하였다.
GBLUP 모형의 적합한 마커 개수보다 적은 수의 참조집단이 존재하더라도 역행렬 GDGV − 1 = (ZBZ′) − 1의 존재는 보장되지 않는다(계산이 되지 않고 바로 다음 단계를 수행할 수 있다.). 이는 마커의 유전자형이 연관불평형 때문에 선형적으로 독립적이지 않거나 동일한 유전자가 나타나는 경우 등이 해당된다. 참조집단의 개체수가 마커수를 초과할 경우, 유전체 행렬은 비변환적(불가역, 비가역)일 가능성이 더 높다. 단, residual polygenic effect를 포함하면 수치의 안정성이 향상될 수 있으며, 다음과 같은 방정식을 도출할 수 있다.

상기 방정식을 사용하여 유전체육종가를 참조집단에 대해 각각 RPG와 DGV로 변환할 수 있다. SNP BLUP 모형을 이용하여 참조집단의 RPG효과를 추정하기 위해 첨자 0으로 표시된 유전체 자료가 없는 선조와 참조집단의 혈연계수행렬은 다음과 같이 정의될 수 있다.

그리고 상기 혈연계수행렬을 역행렬 하면,

분할(partitioned) 행렬의 역행렬 규칙에 따라(Harville, 1997) 다음과 같이 표시된다.

참조집단의 유전체자료가 없는 선조의 RPG에 대한 방정식을 추가함으로써 SNP BLUP 모델에 대한 Henderson의 혼합 모형 방정식은 방정식과 같다. 여기서 â0는 참조집단의 유전체자료가 없는 조상의 RPG를 나타낸다.

Single step SNP model은 Interbull 등에서 사용되고 있으며(Liu et al, 2014) Single step SNP model을 사용하기 위해서는 먼저 유전체 혈연계수행렬의 방법을 고려해야 한다. 현재 Interbull의 유전체 신뢰도 추정 방법은 유전체 개체 수에 대한 제한이 없으며, 모든 참조집단을 포함하는 SNP 관계 매트릭스의 역행렬을 구할 수 있다. 다른 대안들은 역행렬 시 너무 커지게 되는 유전체 혈연계수행렬과 함께 계산되기에 효율성이 떨어진다. Interbull 유전체 신뢰도 추정 방법의 또 다른 특징은 EDC를 얻기 위해 역행렬을 수행할 필요가 없다는 것이다. Interbull 유전체 신뢰도 추정 방법은 Interbull 표준 절차에 따라 계산된 개체들의 EDC를 요구하며, Interbull 유전체 신뢰도 추정 방법은 유전체 검증을 유전체 신뢰도 추정 절차의 일부로 통합함으로써 실현되며, 유전체의 신뢰성에 근사치를 두고 계산하게 된다.
본 연구에서는 상기 방법을 기반으로 자체 개발된 Fortran 프로그램은 SNP BLUP REL을 사용하였다. 유전체 신뢰도 추정에 대한 논문에서는 genomic 평가에 사용되는 것과 동일한 allele frequencies를 SNP BLUP 신뢰도 추정 소프트웨어로 권장하고 있다.
③ 유전모수
본 연구에서 사용된 유전모수 값을 Table 1에 제시하였다. 사용된 유전모수 값은 손(2020)의 결과를 이용하였다. 도체중, 등심단면적, 등지방두께 및 근내지방도의 유전분산 값은 각각 500.8±47.01, 29.92±3.06, 8.738±0.85 및 1.545±0.26, 잔차분산 값은 각각 1,298.5±31.01, 77.28±2.82, 15.973±0.50 및 1.662±018, 유전력은 각각 0.28±0.02, 0.28±0.03, 0.35±0.03 및 0.48±0.07로 제시되었다.
|
Table 1. Estimated genetic(σa2)and residual(σe2)variance components, heritabilities(h2)), phenotype genetic correlations among traits of Hanwoo steer 
|
④ 정규성 검정
한우 도체형질의 유전체육종가를 추정하여 추정된 유전체육종가의 정규성을 파악하기 위하여 각 형질별로 분포를 나타내어 정규분포에 근사되는지를 파악하였다. 정규분석은 SAS UNIVARIATE Procedure의 Checking Varialbles for Normality를 이용하여 Kolmogorov-Smirnov 통계량(D)으로 검정하였다. 이러한 적합도 검정으로 지정 분포로부터 관측값이 제대로 작성되었는지를 확인할 수 있는 비모수적 검정이다. Kolmogorov-Smirnov 통계량(D) 추정을 위한 공식은 아래와 같다.

결과 및 고찰
한우농가에서 사육중인 암소를 평가하기 위한 유전체 분석 집단의 구성은 살아있는 암소인 대상개체의 모근 및 도축된 후대의 조직샘플, 후대의 아비 개체의 정액을 한 쌍으로 묶어 수집 후 유전체자료를 생성하였으며, 분석된 내역은 Table 2에 나타내었다. 분석된 6,616두 중 도축 된 개체는 3,091두로 약 47%의 비중을 차지하였다. 이 중, 암소와 거세우는 각각 2,449두 및 642두 있었으며, 분석에 이용된 보증KPN와 살아있는 암소는 각각 275두 및 3,053두 였다. 거세우 197두는 아직 도축이 되지 않아 도축성적을 가지고 있지 않은 것으로 나타났다.한우 암소는 도축월령이 54개월령 내외로 초산일령이 24.4개월령, 임신기간이 280일 전후임을 감안 했을 때 평균적으로 2산에서 3산 정도 후대를 생산한 후 도축된다(손, 2020). Table 2를 보면 유전체자료를 가지고 있는 암소 평균 도축월령이 64.6개월령으로 유전체자료를 가지고 있지 않는 암소 평균 도축월령인 47.4개월령으로 다소 높게 나타났는데 이는 유전체 분석을 위한 암소 집단의 조건으로 혈통의 연결이 잘 되 있으며, 후대를 많이 생산한 개체를 우선 대상으로 지정하였기 때문이다. 도축성적 간 비교를 해보면 유전체 자료가 있는 암소개체의 도체성적은 평균 도체중, 등심단면적, 등지방두께 및 근내지방도가 각각 357.29 kg, 85.18 cm>2, 14.64 mm 및 4.21점으로 나타났으며, 유전체 자료가 없는 암소개체의 도체성적은 각각 352.92 kg, 83.73 cm>2, 13.32 mm 및 4.36점으로 나타났다. 거세우의 도축 시기는 평균적으로 29개월에서 31개월령에 도축 축하하게 되는데, 유전체자료를 가지고 있는 거세우 2,499두의 평균 도축월령은 29.96개월령이며, 평균 도체중, 등심단면적, 등지방두께 및 근내지방도는 각각 458.94 kg, 96.44 cm>2, 13.80 mm 및 6.39점으로 나타났다. 이는 유전체자료가 없는 거세우 1,431,299두에서 도축월령 평균 30.07 개월령과는 큰 차이가 없었으나, 도체중, 등심단면적, 등지방두께 및 근내지방도의 평균성적이 각각 443.51 kg, 93.72 cm>2, 13.62 mm 및 5.94점 보다 다소 높게 나타났다. 자신의 유전체 자료와 도축자료를 가지고 있는 개체는 유전체 자료를 가지고 있는 개체들 6,616두 중에 암소 642두, 거세우 2,449두였다. 유전체 자료를 가지고 있는 개체와 혈통으로 연결 되었지만 자신의 유전체 자료를 가지고 있지않은 개체는 암소에서는 945,566두, 거세우에서는 약 140만두 가량이 되었다. 암소에서는 유전체 자료를 가지고 있는 개체는 그렇지 않은 개체들보다 약 17개월 가량 더 늦게 도축이 되었지만 그 밖의 도체형질에 대한 평균은 큰 차이를 보이지 않았다. 거세우의 경우 출하시기가 수익에 많은 영향을 끼치므로 도축시기는 큰 차이가 없었으며, 암소와 마찬가지로 도체성적에 대한 값도 큰 차이를 보이지 않았다.

|
Table 3. Basic statistics on carcass traits in Hanwoo using genomic data linked to pedigree 
|
|
CW: Carcass weight (㎏), EMA: Eye muscle area (cm2), BF: Backfat thickness (mm), MS: Marbling score. |
한우 암소, 거세우, KPN의 육종가를 BLUP방법과 ssGBLUP방법으로 추정하였을 때 기대되는 육종가를 Table 4.에 나타내었다. BLUP방법으로 추정된 육종가는 한우 암소에서 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 13.90, 3.78, -0.17, 0.63으로 분석되었다. KPN에서는 그보다 높은 22.51, 6.33, -0.58, 0.93을 나타냈으며, 거세우에서는 16.03, 5.29, -0.83, 0.84를 나타냈다. Single Step 방법으로 추정된 유전체육종가는 한우 암소에서 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 3.32, 1.36, -1.04, 0.13으로 분석되었다. KPN에 대해서는 11.96, 4.14, -1.47, 0.48의 값으로 나타났고, 거세우에서는 6.16, 3.20, -1.74, 0.44로 분석되어 유전체육종가와 육종가 간의 차이는 도체중에서 –10.33, 등심단면적에서 –2.23, 등지방두께에서 –0.89, 근내지방도에서 –0.45로 나타났으며, 상대적으로 유전체육종가 값이 육종가 값보다 낮게 나타났다. 한우 암소, 거세우, KPN의 유전체육종가는 상대적으로 BLUP방법으로 추정된 육종가보다 낮게 나타났다. 이러한 결과가 나오는 이유는 유전체육종가의 자료수가 작아 H matrix를 만드는 과정에서 혈연계수행렬에 유전체 자료의 비중보다 혈통자료에 대한 비중이 높게 차지하여 혈통자료를 이용하는 BLUP방법의 EBV 추정치가 더 높은 수치로 추정된다고 사료된다.


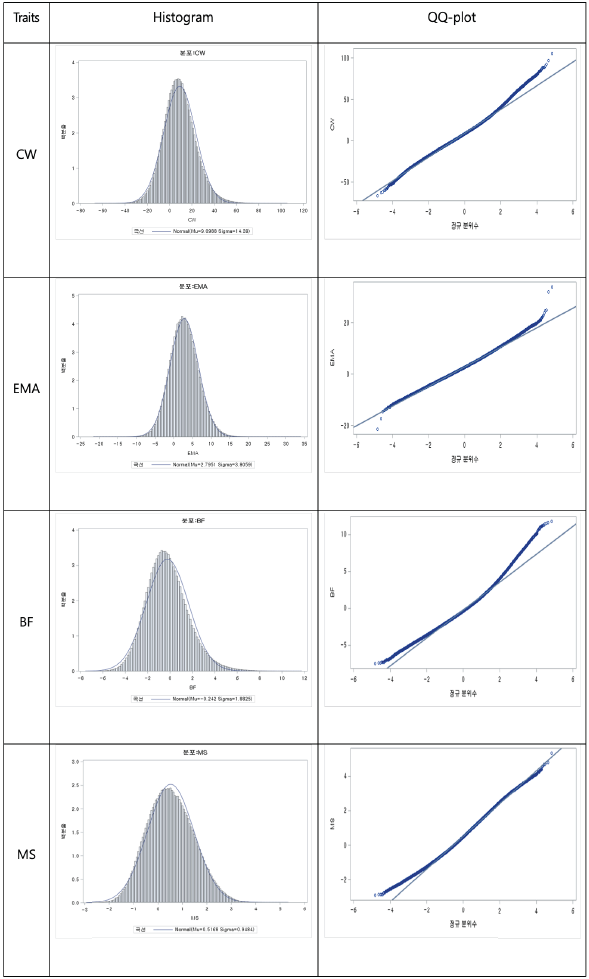
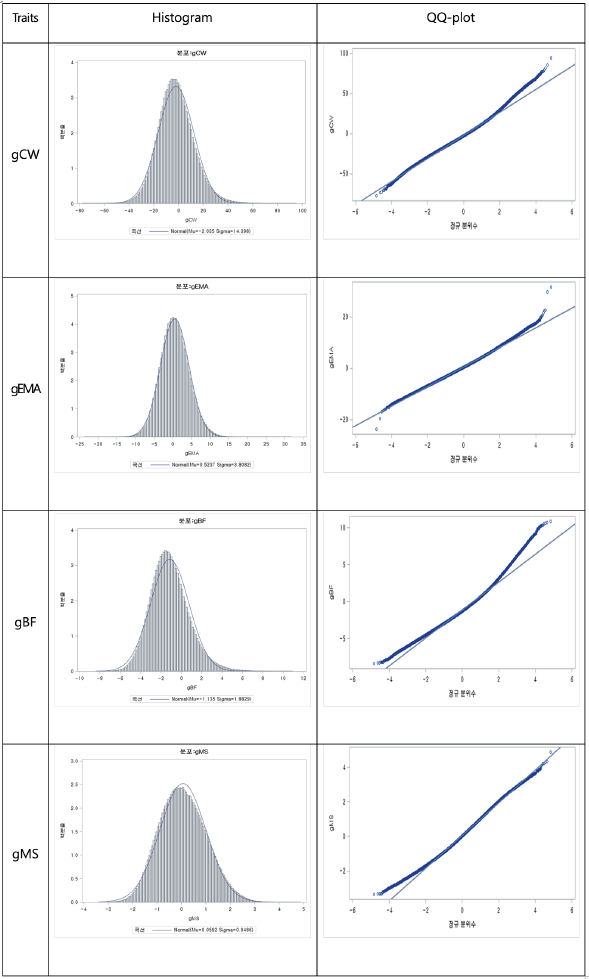
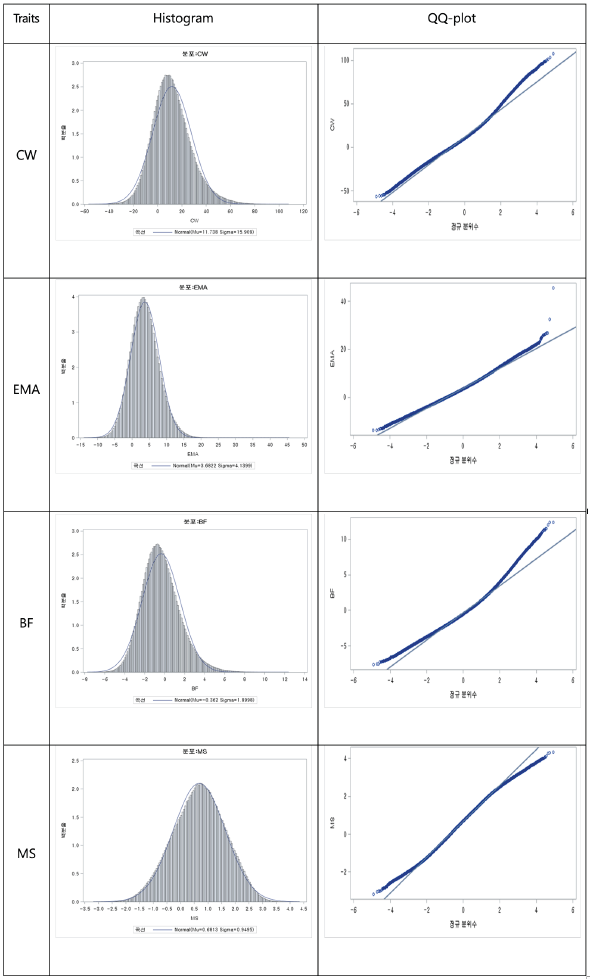
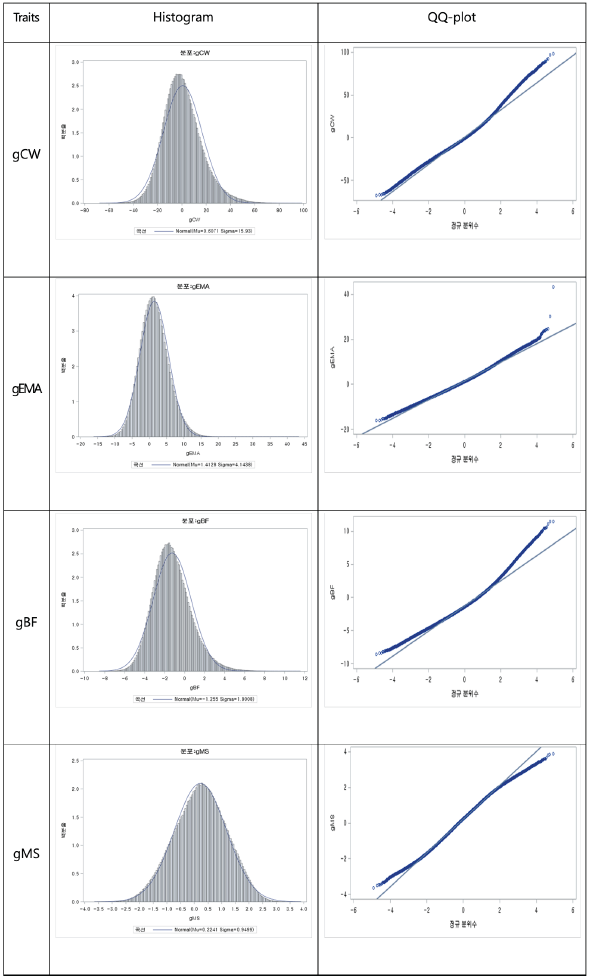
한우 240만두의 개체의 유전체육종가 값이 정규분포를 따르는지 확인하기 위하여 콜모고로프-스미노프(Kolmogorov-Smirnov)검정으로 정규성 검정을 진행하여 Figure 1~4에 제시하였다. 육종가와 유전체육종가에 대하여 정규성을 나타내는지 검정하고자 정규분포함수에 적합시킨 히스토그램 및 QQ-PLOT을 도체형질별로 나타내었다. Figure. 1은 한우 암소의 육종가에 대한 정규성 검정이며 Figure. 2는 한우 암소의 유전체육종가에 대한 정규성 검정 결과이다. Figure. 3은 한우 거세우와 KPN를 전부 수소로 묶어 육종가에 대한 정규성분포를 그린 것이며, Figure. 4도 한우 수소에 대햐여 유전체육종가에 대한 정규성 분포를 그린 것이다. Kolmogorov Smirnov검정한 결과 네 가지 도체형질에 대한 육종가 및 유전체육종가 값은 정규분포를 따르는 것으로 나타났다(P value≤0.05). 유전체 자료를 가지고 있는 한우 6,616두에 대한 도축자료로부터 추정된 육종가의 신뢰도는 암소에서 도체중, 등심단면적, 등지방두께 및 근내지방도 각각 0.48, 0.51, 0.51 및 0.59로 나타났으며 한우 KPN의 추정된 육종가 신뢰도는 0.92, 0.92, 0.93 및 0.94로 평균 40%정도 높게 추정되었다. 반면 Single Step GBLUP방법으로 추정된 유전체육종가 신뢰도는 암소에서 도체중 0.66, 등심단면적 0.68, 등지방두께 0.70, 근내지방도 0.77을 나타냈으며, 한우 KPN에서는 차례대로 0.93, 0.93, 0.94, 0.96으로 나타났다. 한우 암소의 경우 유전체육종가의 신뢰도가 육종가의 신뢰도 보다 평균 17~19% 상승되는 것을 확인하였으며, KPN의 경우 미약하지만 유전체육종가의 신뢰도가 약간 높게 나타났다. 이는 후대의 도체성적이 많이 포함된 KPN의 경우는 육종가 및 유전체육종가에서 높은 신뢰도를 가지고 있으며, 후대의 도체성적이 KPN보다 적은 암소의 경우에는 육종가보다 유전체육종가를 이용한 신뢰도가 높다는 것을 알 수 있었다.