
Introduction
양돈산업에서 등지방 두께는 주요 양적경제형질(quantitative economic trait)로서 정육생산량에 결정적인 영향을 미친다. 등지방 두께가 줄어 들수록 정육생산량이 늘어남으로써 수익률 증대로 이어 질 수 있기 때문이다(Cliplef and Mckay, 1993). 따라서, 양돈산업에서 등지방 두께형질의 유전적 능력 향상은 경제적 효율성과 생산성에 직결되어있다고 할 수 있다.
돈군에서 등지방 두께형질의 유전적 능력 향상을 위해서는 돈군의 유전적 특성을 나타내는 유전력 (heritability, h2) 이나 유전상관계수(genetic correlation coefficient, rg)와 같은 유전모수(genetic paramenter)의 정확한 추정이 필수적이다. 이제까지 경제동물의 유전적 개량을 위한 유전모수의 추정은 가계(pedigree) 정보를 이용하여, 그 가계 구성원 간의 상가적(additive)인 수리적 관계를 계산하여 구축한 혈연관계행렬(numeric relationship matrix, NRM)이 사용되어 왔다(Henderson, 1984). 최근 들어서 유전체학 기법의 급속한 발달로 단일염기서열(single nucleotide polymorphism, SNP) 마커를 고밀도로 집적한 chip 기반의 대용량 유전자형 결정법이 실용화 되었다. 이에 따라 집단(population) 구성원 간 유전적 관계를 SNP마커 정보를 이용하여 SNP chip 기반 유전체관계행렬(genomic relationship matrix, GRM)을 구축할 수 있는 방법들이 개발되었고, GRM은 유전모수 추정을 위한 혼합선형모형 분석에 있어서 NRM을 대치 할 수 있는 대안으로 대두되었다(VanRaden, 2008; Yang 등 2011; Goddard 등 2011).
국민소득증가와 이에 따른 생활 수준의 향상은 돈육소비 패턴의 변화를 가져와 양적인 소비에서 맛과 같은 질적인 소비 위주로 바뀌고 있으며, 이와 함께 양돈산업 및 일반소비자들에게 제주재래흑돼지(Jeju native black pig, JBP)에 대한 관심이 증대되고 있다. JBP는 제주도에서 길러져 오던 재래돼지품종으로써, 요크셔나 랜드레이스와 같은 외래돼지품종에 비하여 적색의 육색, 백색의 단단한 지방과 그리고 우수한 상강도(marbling degree)를 보이지만(Kim 등, 2009), 체구가 작고, 성장이 느리며, 산자수가 적은 단점이 있다. JBP는 경제성이 우수한 외래돼지품종의 제주도 도입으로 멸종의 위기를 맞았지만, 1980년대말 시작된 JBP 복원 및 보존사업으로 순종의 보호와 육성이 진행되어 왔으며, 2015년도에 천연기념물 제550호로 지정되었다. 하지만, 성장형질 및 번식형질에 관련된 단점들로 인해 순종 JBP의 산업화는 아직 미흡한 상황이다. 2006년 JBP를 소재로 이용한 본격적인 산업화를 위해, 단점인 성장형질을 개량하고, 장점인 육질형질을 보존하고자 하는 연구들이 시작되었다. JBP 품종과 랜드레이스 품종 간 F2 교배축군이 조성되어 육질형질에 영향을 미치는 양적 형질 좌위(quantitative trait loci, QTLs)를 전장유전체검색 방법을 적용하여 검출해 낼 수가 있었다(Cho 등, 2015; Yoo 등, 2012). 본 연구에서는 우리나라 재래돼지품종인 JBP와 외래돼지품종인 랜드레이스 품종 간 교배축군(F2세대)에서 얻어진 등지방 두께형질에 대한 유전모수를 SNP chip 및 가계 기반으로 추정하고 이를 비교 분석하였다.
Materials and methods
공시 동물
본 연구에 이용된 F2 집단은 농촌진흥청 국립축산과학원 난지축산연구소에서 JBP와 랜드레이스의 상호교차교배(reciprocal intercross)에 의해서 생산되고 육성되었다(Kang 등, 2016). P세대 돼지들은 랜드레이스가 17두, JBP가 19두가 F1세대 생산에 이용되었다. 상호교차교배를 통하여 생산된 F1세대 91두를 다시 상호교차교배를 통하여 약 1200두의 F2세대 돼지를 생산하였다. 생산된 F2세대 자손 중 총 1,105두(암 537, 수 568)를 유전자형 결정과 표현형 수집에 이용하였다.
분석 자료
모든 F2 세대 돼지들은 평균일령 199일에 동일한 도축장에서 도축되었으며 도축 전에 돼지들은 24시간 공복 시켰으나, 물은 자유급여 하였다. 등지방두께는 좌측 반도체의 세 부위에서 직접 측정하였다. 첫번째 등지방두께는 네번째와 다섯번째 갈비뼈 사이에서 측정하였고(bf_tho4_5, mm), 두번째 등지방 두께는 11번째와 12번째 갈비뼈 사이에서 측정하였으며(bf_tho11_12, mm), 마지막 갈비뼈와 첫번째 요추뼈(first lumbar vertebrae) 사이에서도 측정하였다(bf_tho_lum, mm). 이에 더하여 축산물품질평가원에서 측정한 등지방두께(BF, mm) [(11번째와 12번째 갈비뼈 사이 등지방 두께)+(마지막 갈비뼈와 첫번째 요추뼈 사이 등지방 두께)]/2 도 확보하여 본 연구에 이용하였다. SNP 마커의 유전자형 결정은 PorcineSNP60K BeadChip (Illumina Inc., USA)을 사용하여 수행하였다. 결정된 SNP 마커의 유전자형에 대하여 minor allele frequency(MAF)가 0.05 이하, 유전자형 결측 비율이 5% 이상, 하디-바인베르그평형(Hardy-Weinberg equilibrium)을 극도로 벋어 나는 경우(P<0.000001)를 제외한 돼지 18개 상염색체상의 SNP 마커 39,992개의 분석결과를 이용하였다. 이와 같은 작업은 PLINK 프로그램(Purcell 등, 2007)을 사용하였다.
통계 분석
측정된 등지방 두께 단일 형질들에 대한 유전력을 추정하기 선형혼합모형은 다음과 같다:
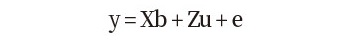 (모형 1)
(모형 1)
여기서,y는 등지방 두께 관측치들에 대한 벡터이고, b는 전체평균, 성별, 배치 그리고 도체중의 고정효과 벡터이며, X는 관측치에 각 고정 효과를 연관시키는 빈도행렬(incidence matrix)이며, u는 개체의 상가적 임의 유전효과 벡터이고, Z는 u에 연관이 된 빈도행렬이고, e는 임의잔차효과 벡터이다. e에 대한 평균과 분산은 e ~N(0, Ise2) 로 설정하였으며, u에 대한 평균과 분산은 u ~N(0, Asu2) 또는 u ~N(0, Gsu2)과 같이 설정하였다. 여기서 I는 단위행렬(identity matrix)이고, A는 혈연관계행렬(NRM)이며(Henderson, 1984), G는 유전체관계행렬(GRM)을 나타낸다. 앞서 얻은 39,992개의 SNP 마커를 이용해서 G를 구하기 위해 SNP 마커의 대립유전자의 빈도와 유전자형을 이용하는 아래와 같은 행렬식이 이용되었다(Yang 등, 2011):
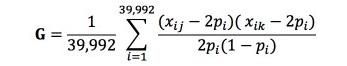
여기서, xij와 xik는 i번째 존재하는 SNP마커에서 j번째와 k번째 개체의 소수대립유전자 개수(0, 1, 2)이며, 는 소수대립유전자의 빈도이다. GRM 구축에는 Yang 등(2011)이 개발한 GCTA(Genome-wide Complex Trait Analysis) 프로그램을 이용하였다. 등지방 두께 형질들간의 유전상관계수를 추정하기 위한 이변량선형혼합모형은 다음과 같다:
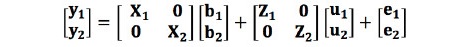
여기서, y1와 y2 는 분석에 고려된 두 표현형에 대한 벡터이고, b1와 b2는 전체평균, 성별, 배치 그리고 도체중의 고정효과 벡터이며, X1과 X2는 표현형관측치에 각 고정 효과를 연관시키는 빈도행렬(incidence matrix)이다. u1와 u2는 두 형질에 대한 개체의 상가적 임의 유전효과 벡터이고, Z1와 Z2는 u1와 u2에 연관된 빈도행렬이고, e1와 e2는 임의잔차효과 벡터이다. 이변량선형혼합모형의 기대치는 다음과 같다.
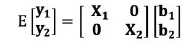
NRM(A)을 사용한 이변량선형혼합모형의 분산은 다음과 같으며,

GRM(G)을 사용한 이변량선형혼합모형의 분산은 다음과 같다.

여기서, σu112, σu122, σu212, σu222 는 고려된 두 형질들의 상가적 유전(공)분산이며, σe112, σe122, σe212, σe222 두 형질의 잔차(공)분산이다. I는 단위행렬(identity matrix)이다.
일변량에 대한 유전분산성분과 그리고 환경분산성분의 추정에는 NRM을 이용한 경우 restricted maximum likelihood (REML) 방법에 기반한 ASReml 프로그램(VSN international, UK)을 이용하였고, GRM을 이용한 경우 genomic restricted maximum likelihood (GREML)방법에 기반한 GCTA프로그램(Yang 등 2011)을 이용하였다. 모형 1을 이용해서 얻은 분산 성분 추정을 위한 결과를 이용해서는 아래의 식을 이용하여 유전력(h2) 및 SNP-유전력(SNP-h2)을 계산하였다:
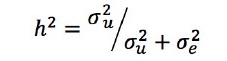
이변량에 대한 유전(공)분산성분과 그리고 환경(공)분산성분의 추정에는 NRM을 이용한 경우 REML 방법에 기반한 ASReml 프로그램(VSN international, UK)을 이용하였고, GRM을 이용한 경우 GREML방법에 기반한 MTG2 프로그램(Lee and van der Werf, 2016)을 이용하였다.
표현형 상관계수 및 유전상관계수는 아래의 식을 이용하여 계산하였다:
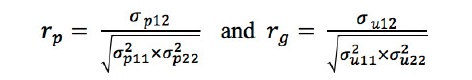
각 모수들의 표준오차는 테일러전개식의 1차 근사(first order Talyor series approximation)를 이용한 델타방법(delta method)을 이용하여 구하였다.
Results and discussion
등지방 두께 측정형질의 기초 통계치들은 Yoo 등의 보고(2012)에 이미 제시되어 있으며, bf_tho4_5, bf_tho11_12, bf_tho_lum 및 BF의 평균은 각각 34.0 mm, 28.0 mm, 26.2 mm, 그리고 22.9 mm 이었다. Porcine SNP 60K BeadChip을 이용하여 유전자형이 분석된 총 62,163개의 SNP 표지들 중에서 quality control 작업을 거친 전체 39,992개 SNP의 상염색체 상의 위치에 분포에 대한 정보는 Park등의 보고(2016)에 제시되어 있다.
Figure 1 에서는 F2 교배축군의 돼지들의 SNP 정보를 이용하여 구성된 GRM을 heatmap으로 나타내었고 GRM의 대각선 원소(diagonal element) 및 비대각선 원소(off-diagonal element)들의 분포를 나타내는 히스토그램이 제시되어 있다. GRM heatmap을 살펴보면 F2 교배축군의 15 전형매(fullsib) 가계를 나타내는 블록들을 관찰 할 수 있다. 이 블록들을 구성하는 개체들간의 genomic relationship은 대략 0.25임을 알 수 있다 (Figure 1a). 대각선원소 분포의 평균은 기대치와 같이 1이었으나 표준편차가 0.085로 계산되어 평균을 중심으로 한 변이가 존재함을 알 수 있다 (Figure 1b). 비대각선원소 분포의 평균은 0에 근사하였으나 역시 상당한 변이가 존재하였다 (Figure 1c).
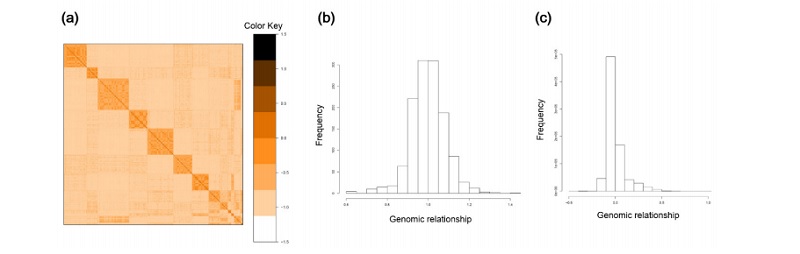
Table 1는 공시축군에서 bf_tho4_5, bf_tho11_12, bf_tho_lum 및 BF에 대한 유전 분산성분과 환경 분산성분, 그리고 유전력을 모형 1을 기반으로 REML과 GREML 방법을 이용하여 추정한 결과를 요약하였다. 모형 1을 이용해서 REML 방법으로 구한 유전력 추정치는 0.48에서 0.57이었며, GREML 방법으로 구한 유전력 추정치는 0.48에서 0.60으로 두 방법사이의 추정치가 거의 유사하였다. 하지만, 이들 추정치의 표준오차를 살펴보면 REML의 경우는 0.10에서 0.11이으나, GREML의 경우는 0.04에서 0.05으로서 REML에 의해 추정된 표준오차에 비해 거의 반 수준을 보였다(Table 1).
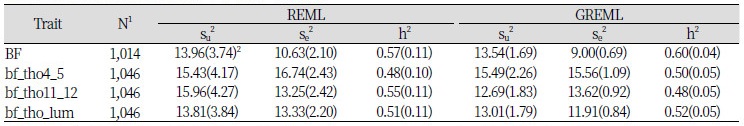
Figure 2는 공시축군에서 네 가지 등지방 두께 형질에 대해서 이 들의 표현형상관계수와 유전상관계수를 추정한 결과이다. 표현형 상관계수의 추정 범위는 0.62에서 0.83으로 고도의 양의 상관관계를 나타냈다. 모형 2에서 REML 방법으로 구한 유전상관계수의 추정치는 0.80에서 0.99로 추정되어 표현형 상관계수 보다 높은 양의 상관관계를 나타냈으나, GREML 방법으로 구한 유전상관계수추정치는 0.93에서 0.99으로 추정되어 더 높은 양의 상관관계를 나타냈다. 이들 추정치의 표준오차를 살펴보면 REML의 경우는 0.01에서 0.09이었으나, GREML의 경우는 0.003에서 0.02로서 REML에 의해 추정된 표준오차에 비해 낮은 수준을 보였다. 이와 같이 높은 수준에서 추정된 양의 유전상관계수는 비록 등지방 두께를 측정한 부위는 다르지만 유사한 유전요인들이 등지방 두께 변이에 영향을 주는 것으로 사료된다.
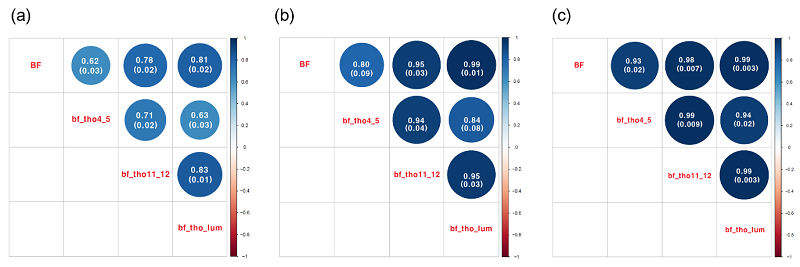
Figure 2 (a) Phenotypic correlation coefficients of the backfat thickness traits. (b) Phenotypic correlation coefficients (REML estimates) of the backfat thickness traits. (c) Phenotypic correlation coefficients (GREML estimates) of the backfat thickness traits. Numbers in parentheses represent standard error of each estimated coefficient.
동일한 통계 모형과 같은 공시동물에서 비롯된 표현형 자료가 사용되었음에도 불구하고, 이상과 같이 REML 방법과 GREML 방법의 유전모수 추정에 있어서 정확도에서 차이를 보이는 이유를 살펴본다면, 기존의 NRM을 이용하는 REML방법으로 분산성분을 추정하게 되면, Mendelian sampling variance이 이용하지 못하게 된다. 예를 들어 전형매(full-sib)의 경우 동일한 정보를 분산성분추정에 제공 할 수 밖에 없다. 반면에, GRM을 이용하는 GREML방법을 이용하게 된다면 Mendelian sampling variance를 이용 할 수 있게 되어 전형매(full-sib)의 경우라도 상이한 정보를 분산성분추정에 제공 할 수 있어 좀 더 정확한 분산성분추정치를 기대 할 수 있다(Hill and Weir, 2011). 하지만, 본 연구에서 사용된 F2 교배축군의 경우 유전체 구조가 이질적(heterogeneous)이므로 추정 된 유전모수의 해석에 조심을 기울일 필요가 있다.
본 연구에서는 제주재래흑돼지와 랜드레이스의 교배축군으로부터 등지방 두께형질의 유전모수를 추정을 위하여 기존의 가계정보를 이용한 방법과 SNP chip 분석에서 산출된 유전자형 정보를 활용한 방법을 비교하였고, 그 차이를 “Mendelian sampling variance” 를 이용 하여 고찰하였다. 본 연구의 결과는 제주재래돼지를 기반으로 계통집단 구축에 있어 등지방 두께형질 형질 개량 연구에 기초자료를 활용될 것으로 기대된다.

