
Introduction
가축의 유전체해독이 완료되고, 유전자형결정 기술이 급격하게 발달하면서 가축에서도 고밀도의 DNA 마커 정보(단일염기변이)를 손쉽게 이용할 수 있게 되었다. 이러한 유전자 마커는 다양한 목적으로 사용되고 있다. 사람, 가축 및 식물 유전학 분야에서 형질과 연관된 양적형질좌위 탐색(QTL mapping)뿐만 아니라 유전변이를 이용한 개체의 유전능력의 예측이 가능해 졌다. 인체유전학 분야에서는 질병에 대한 유전력 및 개인의 위험도 추정등이 활발히 연구되고 있다(Yang et al., 2010). 이러한 유전자검출 및 개체의 유전능력 예측을 위해서, 다양한 통계 방법이 매우 유용하게 활용되어 왔다. 이중에서 단일마커 선형회귀분석(linear regression method)은 유전변이와 표현형간 연관분석과 같이 형질과 연관되어 있는 양적형질좌위(QTL)검출에 매우 유용하게 활용되었다. 그러나, 추정해야 하는 유전변이의 수가 관측된 표현형의 수보다 훨씬 많기 때문에 추정에 대한 통계적인 문제가 존재한다. 그 중에 하나가 각 유전변이(SNP)의 편의추정, 즉 SNP 효과 추정에 있어서의 over-estimation이다. 이러한 문제를 해결하기 위해서 유전변이(SNP)의 효과를 임의효과(random effect)로 적합시켜 각 유전변이의 편의추정의 문제를 어느정도 해결 할 수 있다. 아울러, 비선형모형인 Bayes A, Bayes B [2, 3], 그리고 Bayes C [4]는 일부 게놈 영역이 보다 많은 유전분산을 설명하고 있다고 보고 이 지역에 더 많은 가중치를 적용하여 SNP의 효과를 추정할 수 있다. 즉 특정 염색체 영역에 있는 유전변이가 특정 형질에서 보다 큰 유전분산을 설명하고 있다고 가정한다. 그러나 가축개량에서 주로 활용하는 gBLUP 모델은 모든 유전자좌위에 동일한 유전분산을 부여하고 본질적으로 모든 유전자좌를 균등하게 취급하여 유전자효과를 추정한다. 결과적으로 볼 때, 유전체선발에 대한 모의실험결과(Meuwissen et al., 2001)가 발표된 이후, VanRaden et al. (2009)이 제시한 결과에 따르면 혈통정보를 이용하여 추정한 BLUP 모델보다 유전체정보를 이용한 gBLUP 모델이 약 20-50% 높은 정확도를 보였다. 또한 유우 4,500마리의 유전체정보를 이용하여 추정한 육종가가 부모의 혈통에 기반한 육종가에 비해 16-33% 보다 높게 추정되었다. 아울러, Moser et al. (2009)은 gBLUP 모델과 비선형모형인 베이지언 모델간의 추정육종가의 정확도의 차이가 매우 작다고 하였다. GBLUP 모델에 있어서 추정육종가의 정확도에 대해서 Habier et al. (2007)은 표현형을 갖는 개체들간의 혈연관계가 낮을수록 추정육종가의 정확도가 감소하는 것을 확인하였다. 따라서 gBLUP 모델은 개체와 개체간의 혹은 참조집단과 선발집단간의 혈연관계가 매우 중요한 요소임을 알 수 있다. gBLUP 모델은 모든 유전자좌위에 동일한 유전분산을 부여하고 본질적으로 모든 유전자좌위를 매우 균등하게 취급하여 유전자효과를 추정한다. 그러나 형질에 따라서 매우 큰 효과를 갖는 QTL이 존재 한다면, gBLUP 모델 보다 Bayes B 모델이 좀더 정확한 육종가 추정이 가능하다(Clark et al., 2011). 따라서 가축의 경제형질과 같은 polygenic 형질에 대해서는 gBLUP모델이 매우 효과적인 육종가 추정 방법 중에 하나임을 알 수 있다. 한우에 있어서 유전체육종가의 정확도는 참조집단 2000여두를 이용하여 분석한 결과, 혈통정보를 이용한 BLUP모델은 약 30-32%로 낮았지만, gBLUP모델에서는 37-42%로 약 7-10% 높게 추정되었다.
Methods
Genomic Best Unbiased Linear Prediction (gBLUP) 원리
gBLUP 모델은 SNP-BLUP 모델처럼, 유전체상에 흩어져 있는 유전마커의 분산이 고르게 분포하고 있다고 가정하에, 유전마커 효과를 추정하고, 그 유전마커효과의 총합을 유전체 육종가로 간주 한다. 즉, 원인유전변이와 유전마커가 연관불평형 관계에 있다고 가정한다.
전통적인 BLUP모델은 개체의 혈통정보를 이용하여 개체간 유전적 유사도, 즉 numerator relationship matrix (A-matrix)를 설정하고 이를 이용하여 개체의 유전능력을 평가하는데, gBLUP모델에서는 혈통이 아닌 개체의 유전자형을 근거로 Genomic relationship matrix (G-matrix)를 설정하여 BLUP모델에 적합시킨다(Habier et al., 2007). 이것이 가능한 이유sms 유전체선발을 위한 BLUP모델은 다음과 같이 쓸 수 있을 것이다. XX’ + lamda*I, 여기에서 X는 모든 유전마커효과에 대응하는 개체의 표현형이다. 즉 X 행렬에는 개체의 모든 유전자형이 포함되어 있다. 따라서 XX’은 개체 유전자형의 교찻값, 게놈 사이의 상관으로 표시 할 수 있으며, 이들 요소에는 A-matrix에서 보여주는 상가적 혈연관계행렬과 같은 값이 포함되어 있다. 이에 대해서, Habier (2007)는 만약 연관불평형관계가 존재하지 않더라도, 이 방법을 이용하여 추정하는 유전체육종가의 정확도는 단순히 A-matrix처럼 혈연관계로 추정하는 것과 같이 0이 아닐 것이다. 그러나 모의실험을 통한 결과에서 보면, 단순히 혈연관계를 이용하여 추정한 유전체육종가의 정확도가 세대(generation)가 지나면서 급격하게 낮아지는 것을 확인하였다. 이는 세대가 지나면서, 마커사이의 연관불평형관계가 깨지면서 더 이상 원인변이와 연관되어 있는 마커효과를 추정할 수 없기 때문이다. Habier (2007)의 결과에서 볼 수 있는 또 다른 중요성은 이론적으로, 그리고 컴퓨팅적으로 매우 간단하게 유전체육종가의 추정이 가능하다는 것이다. 한가지 매우 간단하게 유전체육종가를 추정할 수 있는 방법은 기존에 상용화되어 있는 ASReml software (Gilmour et al., 2009)를 이용하는 것이다. 여기에서 “n”개의 참조집단(reference population), 표현형과 유전형을 보유한 animal의 표현형 정보를 참조집단(reference population, n) 및 선발집단(selection population; q)이 포함된 “n + q”의 유전체혈연행렬(genomic relationship matrix)과 합하여 gBLUP 모델을 설정한다. ASReml 소프트웨어는 inverse G-matrix를 혼합모형에 직접 적용할 수 있으며, 이 혼합모형에는 선발집단 개체의 유전공분산(genetic co-variance)행렬을 이용하여 유전체 육종가를 추정한다. 혼합모형 방정식은 아래와 같다(Clark and Vanderwerf, 2013).
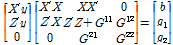
여기에서, 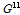 은 참조집단(표현형, 유전자형을 모두 가지고 있는 집단)에 포함된 가축들의 유전체관계행렬을 의미하고,
은 참조집단(표현형, 유전자형을 모두 가지고 있는 집단)에 포함된 가축들의 유전체관계행렬을 의미하고, 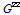 는 선발집단(유전자형만을 보유하고 있는 어린 가축집단)에 포함되어 있는 개체의 유전체관계행렬을 의미한다. 그리고
는 선발집단(유전자형만을 보유하고 있는 어린 가축집단)에 포함되어 있는 개체의 유전체관계행렬을 의미한다. 그리고 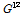 는 참조집단과 선발집단간 유전공분산(genetic co-variance; 다른 개체들간의 유전적 유사도)을 포함하고 있는 유전체관계행렬을 의미한다. 여기에서 중요한 것은 참조집단과 선발집단간의 유전공분산부분(
는 참조집단과 선발집단간 유전공분산(genetic co-variance; 다른 개체들간의 유전적 유사도)을 포함하고 있는 유전체관계행렬을 의미한다. 여기에서 중요한 것은 참조집단과 선발집단간의 유전공분산부분(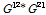 )를 이용하여 표현형을 갖고 있지 않는 선발집단 개체의 유전체육종가 추정이 가능하다.
)를 이용하여 표현형을 갖고 있지 않는 선발집단 개체의 유전체육종가 추정이 가능하다.
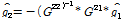 - 육종가 추정 모델식
- 육종가 추정 모델식
이것은 표현형정보가 없는 선발집단 개체의 육종가를 추정하기 위한 회귀계수를 의미한다. 이러한 접근방법을 GBLUP 방법이라고 명명하고, 이는 기존 BLUP 방법과 아주 유사하다. Pedigree BLUP을 이용하여 개체의 육종가를 추정한다면, 아래의 계산식이 될 것이다.
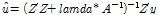
그러나 pedigree BLUP이 아니라, 개체의 유전체정보를 이용한다면, A-matrix를 G-matrix로 대치하여 다음과 같이 추정이 가능할 것이다.
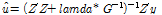
Practice
개체의 유전변이정보를 이용하여 육종가추정 실습
ASREML은 동물유전육종분야에서 BLUP 모델을 이용한 육종가추정에 사용하고 있는 소프트웨어로서 사용하기가 간편하며 확장성이 매우 높다. 가축개량에서 유전체정보를 이용하면서 유전체육종가 추정방법론(GBLUP) 또한 활발하게 개발되고 있다. 이번 장에서는 가축의 유전체육종가 추정방법론 중 참조집단(reference population)에 포함된 개체들의 유전체정보를 이용한 유전체관계행렬(genomic relationship matrix; 유전체정보를 이용한 분산공분산행렬)을 계산하고, 이를 활용하여 표현형이 없는 선발집단(selection candidates) 개체들의 유전체육종가를 추정하는 실습을 하고자 한다. 먼저, ASREML을 이용하여 유전체육종가를 추정하는데는 2가지 단계를 거친다. 따라서, 이를 2 step방법이라 하는데, 첫째로 개체의 유전자형을 이용하여 유전체관계행렬(GRM)을 계산하는 단계와, 둘째로 계산된 GRM을 혼합선형모형에 적합시켜 유전체육종가(GEBV)를 추정하는 단계로 구성되어 있다. 그러나 실제 분석에서는 다른 모든 분석과 마찬가지로 이들 2단계를 수행하는데 훨씬 복잡한 과정이 있어 아래의 모식도에서 개체의 유전자형(유전체정보)을 다루는 과정에 대해 설명하고자 한다.
개체의 유전자형 정보 확보(Genome studio 프로그램 활용법)
개체의 유전자형을 다루기 위하여 사용자의 컴퓨터에 illumina사의 genome studio프로그램을 설치하고, plink plugin 프로그램을 함께 설치하면, 대용량 유전자형 데이터를 plink에서 인식할 수 있는 input 파일을 자동으로 생성할 수 있다. 아래의 그림과 같이 두 소프트웨어를 설치하였으면, genome studio 소프트웨어가 인식할 수 있는 *.bsc 프로젝트 파일을 열고 개체의 유전형을 다운받을 준비를 한다. Genome studio project file (*.bsc)은 아래의 그림과 같이 유전자분석 회사(illumina)로 부터 제공된다.
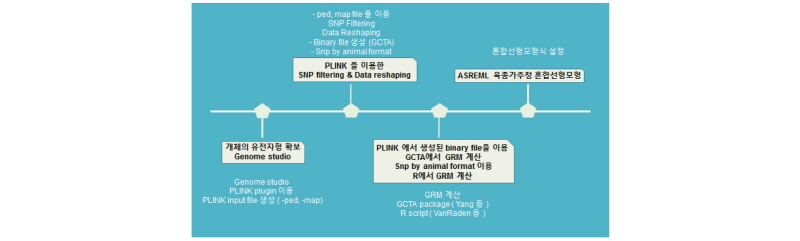
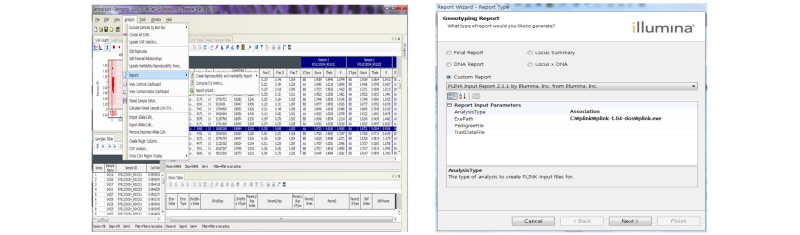
프로젝트파일을 열게 되면, 아래와 같은 화면이 보이게 되고, Analysis 탭에서 Reports 탭을 열게 되면, Report Wizard탭이 보인다. 이 탭을 클릭하면 Genotyping report탭이 열리게 되고 이중에서 Custom report를 클릭하게 되면 Plink plugin이 보이게 된다. 다음(Next)을 클릭하고 Finish를 클릭하게 되면 *.bsc 파일이 있는 폴더에 Plink 폴더가 생성되고 그 안에 Plink input file (-ped, -map)이 만들어 지게 된다.
Plink는 대용량 유전자형자료를 쉽게 다룰 수 있는 유용한 소프트웨어이다. 현재, plink v1.9는 아주 유용한 기능 들이 많이 있다. 특히, 대용량의 SNP 데이터의 필터링 혹은 여러 다른 유전분석 프로그램과 연동할 수 있는 다양한 시작파일(input file)을 쉽게 만들 수 있는 유용한 프로그램이다. Plink 설치는 아래의 홈페이지에서 다운로드 할 수 있다.
http://www.cog-genomics.org/plink2
아래의 스크립트는 간단하게 plink에서 map 파일과 ped 파일을 불러서, SNP 필터링(quality control)을 수행하는 명령어 이다.
|
Plink1.9 --noweb --cow --ped SNP.ped --map SNP.map --hwe 0.001 --maf 0.01 --mind 0.4 --make-bed --out SNP |
명령어를 실행하게 되면, 하디-와인버그평형 P value 0.001 이하, minor allele 1%미만, 그리고 개체의 missing 유전자형이 40%이상인 개체들을 제거한 후에 이진수로 구성된(binary file)을 SNP라는 이름으로 생성하라는 명령어이다. 명령어들에 대한 자세한 설명은 아래 표와 같다.

위의 PLINK 명령어는 위에서 설명한 것과 동일하며, *.ped, *.map파일을 직접 입력하지 않아도 --bfile을 이용하면 *.ped, *.map file 을 자동으로 불러올 수 있다. 그리고, --recodeA는 *.ped 파일의 SNP 유전자형(genome studio top allele, AA, AT, TT)을 additive mode인 0, 1, 2로 재설정하라는 명령어이다. 아래에 제시된 R 스크립트를 이용하여 GRM을 계산하기 위해서는 *.ped 파일의 유전자형(genome studio top allele, AA, AT, TT 등)을 0, 1, 2로 재설정해야 한다. 이때, 최소 대립유전자(minor alleles)는 항상 2로 coding 하는 것이 일반적인 규칙으로, plink의 --recodeA 명령어가 유전자형을 0, 1, 2로 변환을 수행한다.
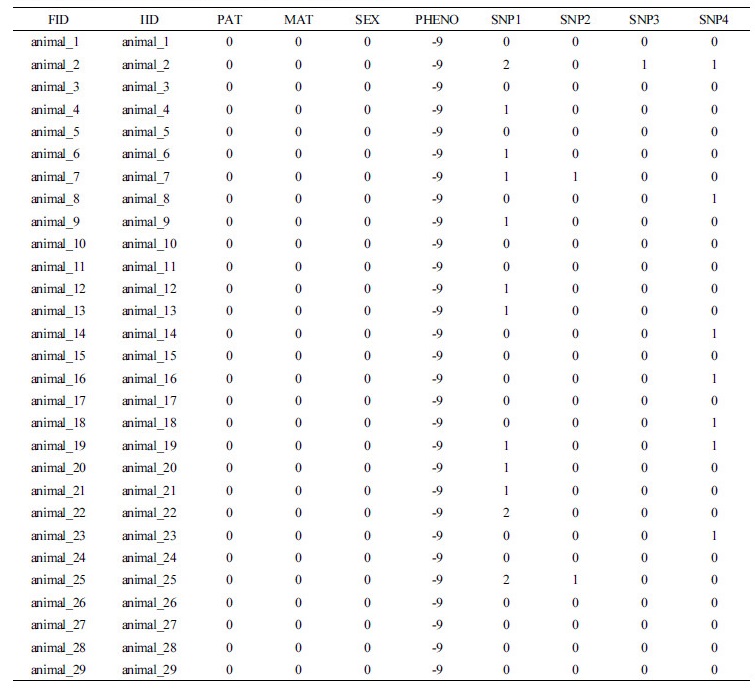
아래에 제시된 R script가 인식하는 유전자형 자료의 구조는 아래의 예시와 같이 행(row)에는 개체정보가 그리고 열(column)에는 유전자형(SNP)형태로 구성되어야 한다(Table 2).
Genomic Relationship Matrix (GRM)계산 (R 및 GCTA 활용법)
R-script를 이용한 GRM 계산
위의 genotype file이 만들어 졌으면, 아래의 스크립트를 이용하여 GRM을 계산할 수 있다. GRM에 대한 구체적인 정보는 Foni 등(2010)이 발표한 논문을 참고하기 바란다. R script를 실행하기에 앞서, script를 실행하기 위한 Package를 아래와 같이 설치한다.
|
source("https://bioconductor.org/biocLite.R") biocLite("GeneticsPed") install.packages("RColorBrewer") |
위의 명령어를 R에서 실행하여 두 개의 Package를 설치한다.
위의 R-스크립트를 이용하여 GRM을 계산하면, 총 3개의 열을 얻을 수 있다. 첫째와 둘째열은 개체 ID, 그리고 셋째열은 GRM을 얻을 수 있다. 이 파일을 열어, 1-3번째 열의 모습을 확인해보기 바란다.
GCTA 소프트웨어를 이용한 GRM 계산
GCTA는 전장유전체정보를 이용한 복잡형질의 유전분석을 위한 소프트웨어로 다음의 홈페이지에서 다운로드하여 설치한다.
http://cnsgenomics.com/software/gcta/download.html
GCTA 프로그램은 대용량자료를 매우 빠르게 분석할 수 있는 장점이 있으며, 유전체연관행렬(genomic relationship matrix, GRM)을 이용하여 복잡형질의 유전력, 유전상관, 유전체육종가추정, 전장유전체연관분석 등 다양한 분석을 동시에 수행할 수 있어, PLINK와 더불어 매우 유용한 분석 패키지이다. 특히, PLINK에서 제공하지 않는 혼합모형(linear mixed model)기반 전장유전체연관분석 기능을 제공하고 있어 분석하려고 하는 집단에 family structure가 존재하거나, 가축과 같이 복잡한 혈통에 의해서 만들어진 집단에서 전장유전체연관분석 및 유전체육종가 추정연구에서 매우 유용하게 활용 할 수 있을 것이다. 위의 홈페이지에서 프로그램을 다운로드하여 설치한 후 사용하면 된다. 특히, GCTA 소프트웨어는 PLINK에서 만들어진 binary file (*.fam, *.bim 그리고 *.bed)을 인식 할 수 있어 대용량 SNP 자료를 PLINK에서 분석용도에 맞게 재설정하여 GCTA 소프트웨어와 연동이 되는 파일을 생성할 수 있다. 즉, PLINK에서 --make-bed 기능을 이용하여 binary file을 생성한 후 GCTA에서 원하는 분석을 할 수 있다. 먼저, 이번 절에서는 GCTA에서 Genomic Relationship Matrix를 생성한 후 이를 활용하여, ASREML에서 유전체육종가 추정에 활용하는 부분을 실습하도록 한다.
|
Plink1.9 --noweb --cow --ped SNP.ped --map SNP.map --hwe 0.001 --maf 0.01 --mind 0.4 --make-bed --out SNP |
위의 명령어는 PLINK에서 초기 genome studio 소프트웨어에서 생산된 SNP.ped, SNP.map 파일을 이용하여, 하디-와인버그(P<0.001), minor allele frequency를 1%, 개체의 missing genotype을 40%이상인 개체를 제거하고, 남은 SNP들을 binary file(--make-bed)로 생성하라고 하는 명령어다. 그러면, PLINK는 자동으로 SNP.bed, SNP.fam 그리고 SNP.bim 파일을 생성한다. 이렇게 생성된 binary file을 GCTA에서 받아서 바로 Genomic Relationship Matrix (GRM)를 생성하는데 사용된다.
위의 명령어를 설명하면 아래의 표(Table 3)와 같다.
위의 GCTA 명령어로 생성된 GRM의 압축파일을 해제하면, 다음과 같은 형태의 GRM (GRM.grm)을 볼 수 있다. GRM.grm 파일을 열면 4개의 열(column)이 있는데, 열 1과 2는 개체의 ID, 그리고 열 3은 GRM을 계산할 때 사용한 SNP의 수 마지막으로 열 4는 실제 개체와 개체간의 유전체혈연정보가 들어있다. 그러나 ASREML에서 사용할 최종 *.grm파일은 개체의 ID인 열 1과 2 그리고 혈연정보인 열 4만이 필요하다. 따라서 Figure 5의 (B)와 같이 최종 *.grm file을 만들어 ASREML에서 유전체 육종가를 계산한다.
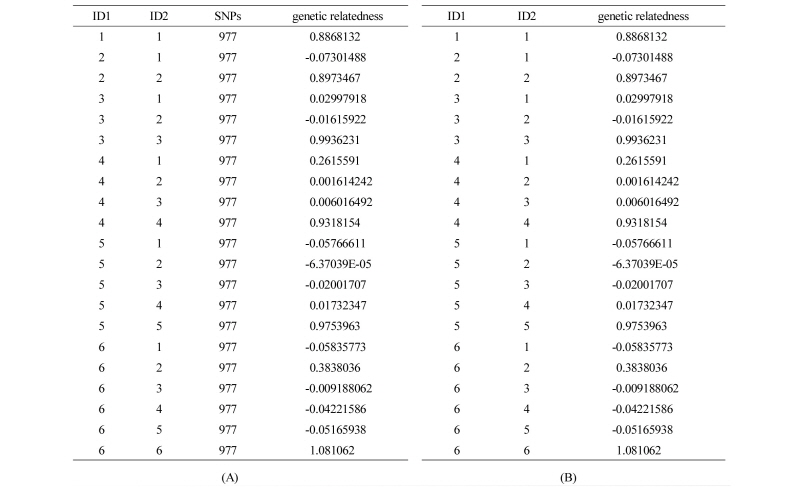
Figure 5의 (A)파일을 (B)파일로 변환을 위해서는 아래의 R-스크립트를 활용하면 쉽게 변환할 수 있다.
|
data<-read.table("GRM.grm.gz",header=F,check.names=F) data2<-data[,c(1,2,4)] write.table(data2,"GCTA_GRM.grm",quote=F,row.names=F,col.names=F,sep="\t") |
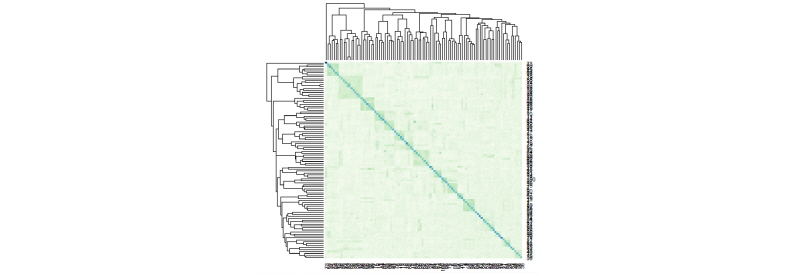
최종적으로 GRM 파일이 생성되면, 아래의 R script를 통하여, 개체와 개체간의 혈연관계를 Heatmap을 이용하여 그려볼 수 있다. 이래의 스크립트를 실행하면, Figure 6과 같은 그림을 생성할 수 있고, 개체와 개체간의 혈연관계를 확인할 수 있다.
ASREML을 활용한 유전체육종가 추정(ASREML 활용법)
ASREML을 이용하여 유전체육종가를 추정하는 방법은 기존 BLUP방법과 동일하다, 다만 A matrix대신 앞에서 추정한 G matrix를 대치하여 준다는 점만 다를 뿐이다. 먼저, ASREML이 인식하는 3개의 파일을 생성해야 한다. 첫째로 *.as 파일이다. *.as 파일에는 ASREML을 실행하라는 명령어들을 넣어야 한다(Figure 8). 그리고 *.as 파일이 인식할 수 있는 표현형이 기록되어 있는 data.txt 파일 그리고 *.grm 파일들을 생성해야 한다. Data.txt 파일에는 Figure 7과 같이, 첫 행에는 animals (개체 ID), qtl(개체순번), 고정효과(year 등) 그리고 마지막으로 표현형으로 구성되어야 한다. 여기에서 열 2의 qtl 번호가 *.sln(결과파일)에서 개체 명호대신 제시됨으로 qtl 일련번호로 개체 명호를 역추적 해야한다.
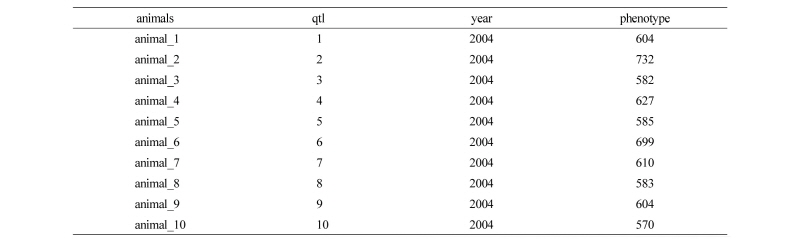

Data.txt 파일과, *.grm 파일, 그리고 아래와 같이 *.as 파일이 설정되면 세 개의 파일(data file, grm file 그리고 *.as file)을 Figure 9와 같이 하나의 폴더에 넣고 asreml을 실행한다.
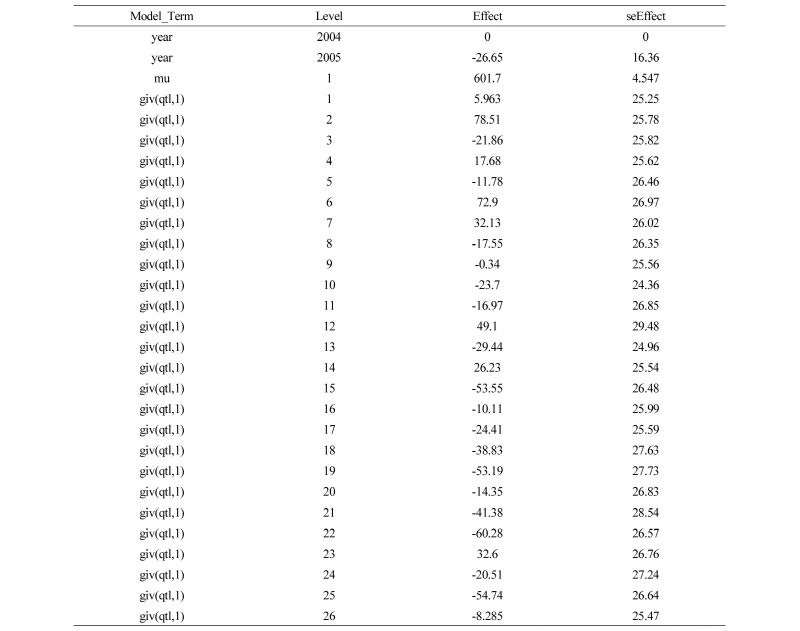
그러면 아래의 결과 파일(*.sln)을 얻을 수 있다. 결과파일(*.sln)은 고정효과에 대한 추정값, 그리고 표현형에 대한 평균(mu)값, 그리고 개체의 육종가 순으로 추정값을 제시해 준다. Effect는 개체의 육종가, 그리고 seEffect는 추정육종가의 오차이다. 여기에서 Level의 숫자(예를들어 1,2,3,4 등)은 data.txt 파일의 qtl의 번호와 일치하고, 이 번호를 이용하여 개체 ID를 추적한다.
요 약
1.GRM (genomic relationship matrix)을 구성할 때 대립유전자의 빈도는 매우 중요하다. 그 이유는 전체적인 matrix의 scale을 조절하기 때문이다. VanRaden이 제안한 최초의 GRM에서는 기초집단의 대립유전자의 빈도를 이용하여 GRM을 구성한다. 그러나, 최근 Forni et al. (2013)에 의하면 base population의 대립유전자의 빈도를 사용하나, 현재 집단의 대립유전자빈도를 사용하여도 추정된 값은 매우 유사하다고 보고한다.
2.ASREM 소프트웨어는 가축개량에서 매우 유용한 프로그램으로 일단 GRM이 만들어 지면 gBLUP모델을 쉽게 설정할수 있는 프로그램이다. 아래의 링크에서 다운로드 할 수 있다.http://www.vsni.co.uk/downloads/asreml
3.ASREML에서 gBLUP을 설정시, GCTA와 같은 소프트웨어를 이용하여 미리 GRM을 구성하여야 한다. 그리고 ASREML에서 육종가 해를 구하기 위하여 inverse를 할 수 있다. GCTA소프트웨어는 아래의 링크에서 다운로드 할 수 있다.http://cnsgenomics.com/software/gcta/#Download

