
Introduction
RNA-seq을 이용한 전사체 분석의 가장 주된 목적은 대조군과 실험군의 차등발현유전자를 확인하고 생물학적 의미를 도출하는 것에 있다. 이를 위해서는 해당 종에 대한 참조전장유전체서열과 유전자정보가 필요하다. 만약, 분석하고자 하는 종의 참조전장유전체 정보가 없다면, RNA-seq 데이터를 드노보 어셈블리를 통해 전사체를 새로 구성하고 드노보 전사체에 RNA-seq 데이터를 정렬해 발현 수준을 확인하는 방법이 있다. 하지만, 드노보 어셈블리 특성상 완전한 전사체를 얻기에는 많은 노력이 필요하며, 해당 드노보 전사체 서열을 이용해 생물학적 정보를 입히는 등의 많은 한계가 있다. 반면, 참조전장유전체 정보가 있다면 RNA-seq 데이터를 직접 정렬해 확인된 모든 유전자의 발현 수준을 예측 할 수 있으며, 더 나아가 novel isoform 전사체를 확인 할 수 있다. 일반적으로 소, 돼지 등 well annotated species의 경우 해당 컨소시움을 통해 유전자의 전장유전체 상의 위치 정보 및 다양한 생물학적 기능 정보를 얻을 수 있다.
차등발현유전자를 선별하는 방법은 실험 설계에 따라 달라진다. 두 그룹간의 일대일 비교 방법과 세 개 이상의 그룹에서 처리 후 시간경과에 따른 시계열 분석 방법, 그리고 여러 조직에서 특이적으로 발현하는 유전자를 확인하는 방법 등이 있다. 각 분석법에는 그에 적합한 통계 모델을 사용해야하며, 두 그룹에서의 분석은 negative binomial 분포모델을 이용한 t-test가 일반적이고, 세 개 이상의 그룹에서의 분석은 pearson correlation과 같은 상관계수를 이용한 K-means clustering 분석 접근법이 가능하다.
통계분석에서 추가적인 반복 데이터를 사용하면 신뢰성이 높고 정확도가 높은 결과를 얻을 수 있다. RNA-seq 분석에서의 반복 데이터는 biological replicate와 technical replicate가 있으며, biological replicate는 여러 개체에서 동일한 실험을 통해 얻은 데이터를 의미하고 technical replicates는 하나의 개체에서 반복적인 실험을 통해 얻은 데이터를 의미한다(Blainey et al., 2014). 많은 연구에서 차등발현유전자 탐색은 반복 데이터가 많고 RNA-seq 데이터 생산량이 많을 수록 효과적이라는 결과가 있었다(Schurch & Nicholas et al., 2016, Liu et al., 2013). 하지만, (conesa & Ana et al., 2016)등은 재현성과 서열해독비용, 그리고 실험 사이의 변동성과 개체간의 변동성을 고려했을 때 각 그룹당 필요한 최소한의 replicate 개수는 3개라는 결과를 보여주었다.
차등발현유전자 분석을 위한 프로그램으로는 Cuffdiff, edgeR, DESeq, Limma 등이 있다. 유전자의 발현 수준을 서로 비교하기 위해서는 RNA-seq 생산량 등의 편차를 보정해야 한다. 프로그램들 간에는 이를 보정하는 방법의 차이가 있으며, cuffdiff는 전체 유래 리드 수(mapped read count)와 유전자 길이를 이용해 보정하는 FPKM(Fragments Per Kilbase of exon per Million fragments mapped)방법을 사용한다. 반면, edgeR은 보정되지 않은 유래 리드 수를 이용해 TMM(Trimmed Mean of M-values) 방법으로 보정한다(Table 1)(Seyednasrollah et al., 2013). 유전자 별 유래 리드 수를 계산해주는 대표적인 분석 도구로는 HTseq(Anders et al., 2015)이 주로 이용된다(Rapaport & Franck et al., 2013).
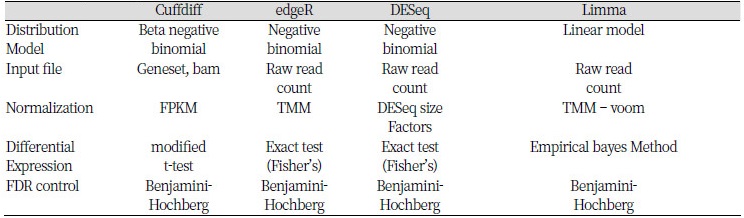
각각의 프로그램들은 사용하는 통계 모델이나 비교를 위한 유전자 발현 수준의 보정 방법, 차등발현유전자 탐색에 쓰이는 통계적 방법 등 여러가지 면에서 차이를 가지고 있기 때문에 실험 목적에 맞는 프로그램을 선별해 사용하는 것이 중요하다. edgeR, DESeq, Limma 등 11가지 프로그램 비교에서 Limma가 여러 조건에서 가장 좋은 성능을 보여주고 이상치(outlier)에 영향을 많이 받지 않으며 분석 시간이 빠르다는 연구결과가 있었다. 또한, DESeq과 edgeR은 비슷한 원리와 비슷한 정확도를 가지고 있음에도 FDR control에 좋지 못하며, DESeq이 edgeR보다 더 보수적인 결과를 보인다(Soneson et al., 2013). 앞서 언급한대로, 차등발현유전자 탐색에 있어서 통계적으로 충분한 검증을 하기 위해 각 그룹당 최소 3개의 샘플이 필요하다. 샘플 수가 적은 경우 Limma와 DESeq 패키지가 좋은 정확도를 보이지만, edgeR은 큰 변동성을 보인다. Cuffdiff는 샘플 수에 큰 영향을 받지 않는다(Seyednasrollah et al., 2013).
Methods
전장 전사체 서열 해독 및 전처리
RNA-seq 데이터 생산을 위한 실험은 준비된 샘플에서 RNA를 추출하는 과정부터 시작된다. 일반적으로 total RNA 2ug정도로 부터 oligodT를 이용해 mRNA를 분리하고 Illumina사의 TruSeq RNA Sample Prep Kit을 이용하여 Library를 제작한다. Library는 일반적으로 100bp Paired-end sequencing을 위해 준비한다. 분리된 mRNA는 fragmentation 단계를 거치고 random hexamer priming을 통해 single-stranded cDNA로 합성된다. 이를 주형으로 second strand가 합성되면 double stranded cDNA가 되며, Blunt-end를 만들기 위한 End Repair, Adapter를 붙이기 위한 A-tailing 및 Adapter ligation을 순차적으로 거친 후, PCR(Polymerase Chain Reaction)을 이용하여 cDNA library를 증폭시킨다. 최종 산물은 2100 BioAnalyzer를 이용하여 확인한다. 만들어진 library 는 KAPA library quantification kit 을 이용하여 정량 한 후 cluster generation하여 Hiseq2500을 이용해 서열 해독을 진행한다. 생산된 RNA-seq 데이터는 서열 해독과정에서 생긴 잘못된 서열을 가진 리드(Read)나 어댑터 서열을 제거하여, 참조전장유전체 서열에 정렬할 때 발생할 수 있는 오류를 최소화해야한다.
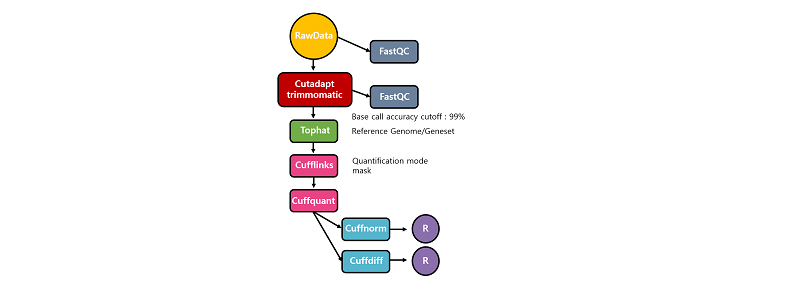
차등발현유전자 탐색을 위한 Tuxedo protocol 분석 개념
Tuxedo protocol 분석방법은 TopHat과 bowtie를 이용해 RNA-seq 데이터를 참조전장유전체에 정렬하고 Cufflinks package를 사용해 차등발현유전자를 탐색하는 일련의 과정을 말한다(Cole Trapnell et al., 2012). TopHat은 alternative splicing 을 고려한 RNA-seq데이터 전용 정렬 프로그램으로 Bowtie를 이용해 짧은 리드들을 매우 빠르게 참조전장유전체에 정렬한다. 해당 정보는 Cufflinks package에서 참조전장유전체의 전사체 영역을 정의하고 발현량 추정을 통해 차등발현유전자를 탐색하는데 사용된다.
Cufflinks package에 포함된 Cufflinks와 Cuffmerge는 전사체 영역 정의를 위해 두가지 분석 목적에 따라 달리 사용된다. 참조전장유전체상의 전사체 영역 정보가 잘 정의되어 있는 종의 경우, novel isoform에 의한 질병 원인 분석 등과 같이 특별한 목적이 아니라면 새롭게 전사체 영역 정보를 정의할 필요가 없으므로, 주어진 gene set 정보만을 이용해 유전자 발현을 정량화한다. 반대로, novel isoform 정보가 필요한 분석에서는 새로운 alternative splicing site정보를 샘플 단위로 확인한 후, 분석에 사용된 모든 샘플의 novel isoform을 하나의 기준으로 합쳐야한다. 이 과정은 차등발현유전자 탐색에서 동일한 기준으로 계산된 발현량을 얻기 위해 필수적이다. 계산된 발현량은 Cufflinks package의 Cuffdiff를 통해 차등발현유전자를 탐색하는데 사용된다.
유전자 발현 정량화 단위 : RPKM (Read Per Kilobase of transcript per Million mapped reads)
RNA-seq 데이터를 이용해 유전자의 발현량을 예측하는 기본적인 방법은 해당 유전자에서 유래된 RNA-seq 리드 수를 확인 하는 것이다. 유래 리드 수 (mapped read count)는 이후 한 개체내 유전자 발현량 비교 및 샘플 간 유전자 발현량 비교에 사용된다. 샘플 간 비교시, 서로 다른 RNA-seq 생산량 등의 원인에 의해 유래 리드 수에 편차가 발생한다. 따라서, 이를 보정해줄 필요가 있다.
RNA-seq analysis에 주로 사용되는 normalization 방법으로는 RPKM, TMM 등이 있으며, Cufflinks 패키지에서는 RPKM을 사용한다. RPKM은 전체 유래 리드 수와 유전자의 길이로 해당 유전자 유래 리드 수를 보정하는 것으로, 샘플 간 비교와 샘플내 유전자 비교 시 발생할 수 있는 편차를 보정한다. 비슷한 방법으로 FPKM(Fragment Per Kilobase of transcript per Million mapped reads)은 생산된 RNA-seq이 paired-end 리드인 경우에 하나의 fragment에서 유래된 두개의 리드임으로 하나의 리드로 계산하는 차이가 있다. RPKM을 이용한 유전자 발현량은 아래의 식을 이용해서 구할 수 있다.
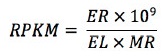
위의 식에서 ER은 유전자에 mapping된 read의 개수, EL은 유전자 길이, MR은 mapping된 read의 전체 개수를 의미한다(Mortazavi et al., 2008). RPKM은 실제 유전자의 발현 유무를 판단할 수 있는 기준으로 활용할 수 있다. RPKM값과 실제 유전자 발현 유무의 상관을 확인한 연구에서 보면(Ramsköld et al., 2009), RPKM값 0.3이상을 갖는 유전자들은 in vivo에서 실제로 발현한다고 판단 할 수 있다.
Cuffdiff를 이용한 차등발현유전자 탐색 통계 테스트 원리
비교 그룹에서 차등발현유전자를 탐색하기 위해서는 측정된 발현량을 이용한 통계적 분석이 필수적으로 필요하다. Cuffdiff는 RPKM값을 이용하여 두 그룹 사이에서 관찰 된 변화에 대한 통계 테스트를 한다(Trapnell & cole et al., 2012). 그룹별로 유전자에 대한 모든 샘플들의 RPKM 평균값을 이용해 유전자에서 각각 t-test를 진행한다. t 통계량 값은 아래의 식을 이용하여 구한다.
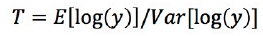
위의 식에서 y값은 두 그룹 사이의 정규화 된 리드 수(normalized count)의 비율을 나타내며, 차등발현유전자 탐색은 t-test를 통해 도출된 p-value값을 이용한다. 전체 유전자에 대해 t-test를 반복 수행 할 시 다중검정으로 인해 유의적으로 차이가 나지 않는 유전자를 유의적인 차이가 있다고 판단 하는 False Positive값이 증가하게 된다(Benjamini et al., 1995). 이 경우, Cuffdiff는 p-value값을 Benjamini-Hochberg방법을 사용해 보정해준다(Rapaport & Franck et al., 2013).
Practice
차등발현유전자 탐색을 위한 실험 설계
본 실습에서는 소(bos taurus)의 RNA-seq 예제 데이터를 이용해 대조군과 실험군의 유전자발현차이를 확인하는 과정에 대해 설명하고자 한다. 실습에 이용될 대조군과 실험군에는 3개씩의 biological replicates을 사용했다. RNA-seq 데이터 생산은 Illumina 사의 TruSeq RNA Sample Prep Kit을 이용해 Library가 제작됐으며, 100 bp Paired-end 로 해독됐다. 데이터는 전처리과정을 거쳐 Tuxedo protocol에 따라 실습에 사용될 것이다.

분석 프로그램 선택 및 설치
RNA-seq 분석을 하기 위해서는 다양한 분석도구들이 필요하다. 본 실습에서는 RNA-seq 데이터에 대한 퀄리티 확인 및 전처리를 수행하기 위해 FastQC, Cutadapt, Trimmomatic을 사용하며, 차등발현유전자 탐색은 Tuxedo protocol을 따라 Tophat, bowtie, Cufflinks package를 이용해 진행할 것이다. 이후, 데이터 시각화는 R이 이용된다.
Bowtie, Cufflinks와 같은 프로그램은 해당 홈페이지에 접속하여 파일 다운로드 경로를 찾을 수 있으며, 서버에 다운로드하고 압축만 풀면 사용할 수 있다.
cd ~
mkdir MyTools
wget [복사된 경로]
unzip bowtie2-2.2.3-linux-x86_64.zip
Samtools, Tophat과 같은 소스코드는 해당 홈페이지에서 소스 코드를 복사하여 서버에서 다운로드한다. Tophat을 설치할 때에는 mapping engine인 bowtie와 호환이 가능한 버전을 사용해야한다.
cd ~/MyTools
wget [복사된 경로]
tar xfj samtools-1.5.tar.bz2
cd samtools-1.5
make
pwd
Cutadapt는 소스코드로 설치할 수 있지만 패키지 관리자를 이용하여 관리자 권한으로 설치할 수도 있다.
sudo pip install cutadapt
sudo pip install –upgrade cutadapt
pip uninstall cutadapt
conda install –c bioconda cutadapt
참조 파일 다운로드 및 인덱스 생성
현재 연구자들이 많이 사용하고 있는 바이오 데이터 베이스에는 NCBI, ENSEMBL, UCSC 등이 있다. 이 중에서도 ENSEMBL은 데이터 큐레이션이 잘 되어있고, 단백질에 대한 정보를 폭넓게 제공하여 유전자 온톨로지와 annotation에 유용하게 이용된다.
참조 파일로는 참조전장유전체서열과 유전자정보를 담고 있는 gene set파일이 필요하며 두 파일 모두 ENSEMBL 홈페이지에서 다운로드할 수 있다. 참조전장유전체서열은 전체 크로모좀의 서열 정보를 담은 파일로 ftp 페이지에 접속하여 wget 명령어를 통해 리눅스서버에 직접 Bos_taurus.UMD3.1.dna.toplevel.fa.gz 파일을 다운로드한다. 저장된 참조전장유전체서열 파일은 reference mapping시 tophat이 이용할 수 있도록 bowtie2와 samtools를 이용하여 인덱스화 해야한다. 이 때, bowtie2는 Tophat의 mapping engine으로 호환이 가능한 버전을 사용한다. Bowite2-build 결과로 *.fa.(1~4).bt2, *.fa.rev.(1,2).bt2 6개의 바이너리 파일이 생성되고 samtools 결과로 *.fa.fai 파일이 생성되며 이 파일은 fa파일에 대한 통계치를 나타낸다.
mkdir Reference
cd Reference
wget [ftp다운경로]/Bos_taurus.UMD3.1.dna.toplevel.fa
bowtie2-build Bos_taurus.UMD3.1.dna.toplevel.fa
Bos_taurus.UMD3.1.dna.toplevel.fa
samtools faidx Bos_taurus.UMD3.1.dna.toplevel.fa
유전자 정보는 gene set에서 GFF3 포맷이나 GTF 포맷인 파일로 다운로드할 수 있다. GFF3와 GTF를 비교하였을 때, GTF는 GFF3보다 정형화된 형식의 유전자 annotation파일이다. 본 분석에서는 GTF 파일을 이용하였다. Gene set 파일은 ENSEMBL홈페이지의 ftp download 페이지를 통해 다운로드할 수 있다.
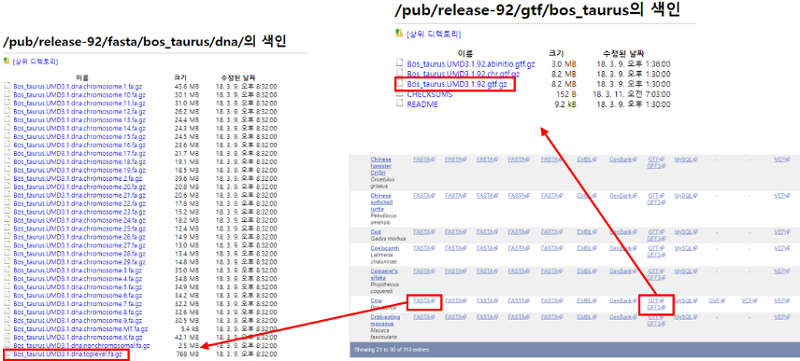
Gene set파일에는 분석하고자하는 mRNA coding 유전자뿐만 아니라 small RNA와 같은 non coding 유전자에 대한 정보도 담고있다. 따라서, 리드가 non-coding 영역에 mapping하여 발생하는 바이어스(bias)를 감소시키기위해 gene set 파일에서 non-coding 영역을 따로 저장한 파일을 생성해야한다. Non-coding 영역 추출은 GTF 파일의 attribute부분 중 gene_biotype에서 protein_coding으로 되어 있는 부분을 제외하고 *.mask.gtf로 저장한다. 이 파일은 유전자 발현량 계산에서 cufflinks의 정확한 발현량 산출을 위한 목적으로 사용된다.
mkdir GeneSet
cd GeneSet
wget [ftp다운경로]/Bos_taurus.UMD3.1.92.gtf
cat Bos_taurus.UMD3.1.92.gtf | grep –v "protein_coding" > Bos_taurus.UMD3.1.92.mask.gtf
RNA-seq 데이터의 퀄리티 확인 및 전처리
RNA-seq 데이터를 참조전장유전체서열에 mapping하기 전에 High throughput sequencing 과정에서 발생될 수 있는 바이어스의 존재 여부와 외래 DNA로부터 유래된 오염으로 인한 문제점을 확인해야 한다. FastQC 소프트웨어(Version 0.11.7)를 이용하여 데이터의 퀄리티를 확인하는 데에 Phred Quality score를 이용한다. Phred Quality score는 Q = -10logP (P = base-calling error probabilities)로 계산되며 예를 들어 이 값이 10일 경우 잘못된 base call의 확률이 100개 중 1개인 것을 의미하므로 90%의 정확도를 나타내어 신뢰할 수 있는 퀄리티를 갖는 서열을 확인할 수 있다. RNA-seq 데이터인 *.fastq.gz 파일을 입력 파일로 사용하며 명령어는 아래와 같다.
~/MyTools/FastQC/fastqc -f fastq --extract -t 8 00.Rawdata/con/bovine_con_1_1.fastq.gz
~/MyTools/FastQC/fastqc -f fastq --extract -t 8 00.Rawdata/con/bovine_con_1_2.fastq.gz

위의 명령어를 실행하면 파일 당 결과 디렉토리와 fastqc_report.html 파일이 생성되고 fastqc_report.html를 통해 총 11가지의 퀄리티 분석 결과를 그래프로 확인할 수 있다. Per Base Sequence Quality는 각 서열에서 베이스 당 퀄리티 값을 보여주며 이 값은 초록색(Pass), 노랑색(Warning), 빨간색(Fail)을 기준으로 boxplot을 통해 나타난다. Per Base Sequence Quality를 통해 quality trimming 전후의 결과를 비교해보면 trimming 이후에 퀄리티가 좋아진 것을 확인할 수 있다. 또한, Per Sequence GC Content는 각 서열의 전체 길이에 대한 GC 함량과 GC 함량의 정규분포 그래프를 비교하여 나타내며 GC 함량은 종마다 다르며 이를 비교해서 데이터의 신뢰성을 검토할 수 있다.
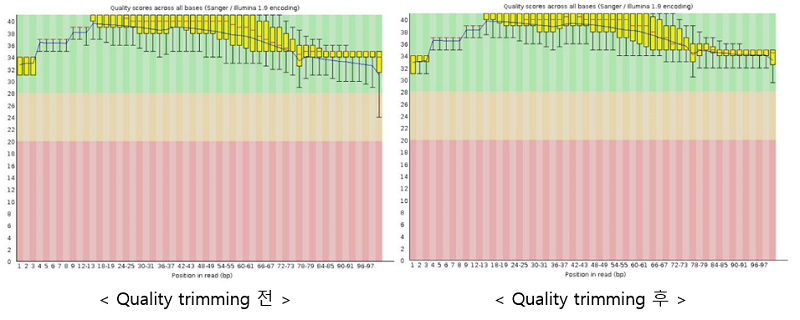
Adapter trimming은 데이터 생성시 삽입 서열(insert sequence)에 남아있을지도 모르는 adapter를 제거하기 위한 과정이다. 본 데이터에 사용된 Adatper는 TruSeq Universal Adapter로 forward기준 3’-end에 GCTCTTCCGATC(T)(12bp)를 가지고 있어서 상보적인 서열인 AGATCGGAAGAGC가 나오기 시작하면 그 이후는 제거된다. 이 과정을 통해 adapter를 제거하고 리드를 참조전장유전체서열에 mapping을 하면 더 정확한 FPKM값을 얻을 수 있다. 퀄리티를 확인한 결과 파일을 입력 파일로 사용하여 Adapter가 제거된 서열이 있는 *.nonadapt.fq.gz 파일을 얻을 수 있다.
mkdir -p 01.Clean/con
cutadapt -a AGATCGGAAGAGC –A AGATCGGAAGAGC
-o 01.Clean/con/bovine_con_1_1.nonadapt.fq.gz
-p 01.Clean/con/bovine_con_1_2.nonadapt.fq.gz
00.Rawdata/con/bovine_con_1_1.fastq.gz
00.Rawdata/con/bovine_con_1_2.fastq.gz

Quality trimming은 Trimmomatic(version 0.38) 프로그램을 이용하여 신뢰할 수 있는 퀄리티를 나타내는 서열을 확인하고 기준 이하의 서열을 제거하기 위한 과정이다. Adapter가 제거된 *.nonadapt.fq.gz 파일이 입력 파일로 사용되며 Phred Quality score를 기준으로 기준 이하의 서열을 제거한다. 그 결과로 아래 명령어를 따르면 base call 정확도가 99%인 서열을 얻을 수 있다.
java -jar MyTools/Trimmomatic-0.36/trimmomatic-0.36.jar PE
01.Clean/con/bovine_con_1_1.nonadapt.fq.gz
01.Clean/con/bovine_con_1_2.nonadapt.fq.gz
01.Clean/con/bovine_con_1_R1.qt.P.fastq.gz
01.Clean/con/bovine_con_1_R1.qt.U.fastq.gz
01.Clean/con/bovine_con_1_R2.qt.P.fastq.gz
01.Clean/con/bovine_con_1_R2.qt.U.fastq.gz
SLIDINGWINDOW:4:20 MINLEN:36
참조전장유전체 정렬을 통한 유전자 발현량 추정
참조전장유전체 정렬 과정은 RNA-seq 데이터를 참조 서열에 정렬하기 위해 TopHat 프로그램을 이용한다. 이는 RNA-seq 리드를 인덱스화된 참조 서열에 정렬하는 과정이며, reference mapping이라 부른다. Quality trimming의 결과 파일 중 *.P.fastq.gz 파일과 참조전장유전체서열 파일을 입력 파일로 사용한다.
mkdir -p 02.Align/con
tophat -o 02.Align/con/bovine_con_1
-p 8
-r 250
--mate-std-dev 50
--library-type fr-firststrand
-G GeneSet/Bos_taurus.UMD3.1.92.gtf
--rg-id control
--rg-sample bovine_con_1
Reference/Bos_taurus.UMD3.1.dna.toplevel.fa
01.Clean/con/bovine_con_1_R1.qt.P.fastq.gz
01.Clean/con/bovine_con_1_R2.qt.P.fastq.gz
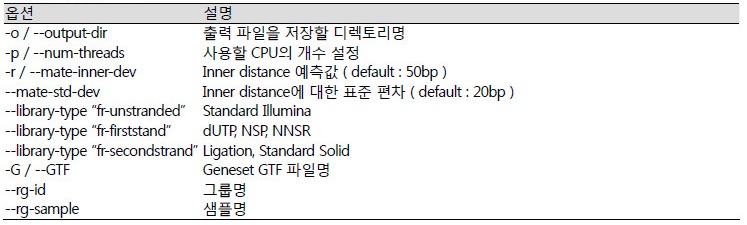
Inner distance는 Insert size에서 양쪽 리드의 길이를 제외한 거리로 계산할 수 있다. 리드가 mapping될 때, 방향성이 다른 유전자가 겹쳐진 부분에서 리드들이 중복되어 mapping이 될 수 있다. 이 경우에 리드의 방향성이 잘못 되어있으면 중복된 리드들에 의해 RPKM값의 정확도가 감소한다. 그래서 Illumina에서는 dUTP방법을 이용하여 리드가 본래 유전자와 방향성이 같도록 만들수 있는 TruSeq Stranded mRNA Library prep. Kit을 제공하고 있으며, library-type 옵션으로 fr-firststand를 이용한다.
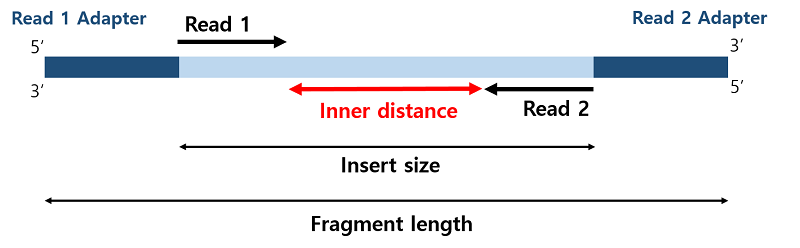
위의 명령어를 실행하면 결과 파일로 accepted_hits.bam, align_summary.txt, deletions.bed, insertions.bed, junctions.bed file이 생성된다. accepted_hits.bam은 리드들을 mapping한 결과 파일이며 align_summary.txt는 reference mapping 통계치가 저장된 파일이다.
Cufflinks 과정에서는 모든 유전자의 발현 수준 예측, 전사체 정렬, novel isoform 모델링을 위해 진행되며 샘플 간의 비교를 위해 normalization을 한다. Cufflinks 패키지에는 Cufflinks, Cuffquant, Cuffnorm이 있으며 이는 발현량을 예측하고 normalization하는 일련의 과정이다. 입력 파일로는 Tophat의 결과 파일인 accepted_hits.bam 파일과 gene set 파일이 사용된다. gene set 파일을 사용하는데 있어서 novel isoform 전사체 확인 유무에 따라 분석 모드가 두가지로 나뉘며, Quantification 모드는 주어진 gene set을 그대로 유전자 발현량을 예측하기 위해 이용한다. 반면, novel isoform 탐색 모드는 주어진 gene set을 기반으로 새로운 novel isoform을 찾기 위해 이용된다.
mkdir -p 03.Cufflinks/con
cufflinks -o 03.Cufflinks/con/bovine_con_1
-p 8
--library-type fr-firststrand
-G GeneSet/Bos_taurus.UMD3.1.92.gtf
--mask-file GeneSet/Bos_taurus.UMD3.1.92.mask.gtf
--frag-bias-correct Reference/Bos_taurus.UMD3.1.dna.toplevel.fa
02.Align/con/bovine_con_1/accepted_hits.bam

Cufflinks의 결과로 genes.fpkm_tracking, isoforms.fpkm_tracking, skipped.gtf, transcripts.gtf 파일이 생성되며 genes.fpkm_tracking 파일은 유전자 발현량에 대한 FPKM값을 나타낸다. isoforms.fpkm_tracking 파일은 예측된 isoform 수준 발현량을 나타내고, transcripts.gtf은 정렬된 isoform을 갖는 GTF 파일이다.
Cuffquant 과정은 각 샘플별 유전자와 전사체의 발현량을 확인하고 CXB 포맷으로 결과 파일을 저장하는 과정이다. Tophat의 결과 파일인 accepted_hits.bam 파일을 입력 파일로 사용하며 분석 결과로 abundances.cxb 파일이 생성된다.
mkdir -p 05.Cuffquant/con
cuffquant --output-dir 05.Cuffquant/con/bovine_con_1
--num-threads 8
--library-type fr-firststrand
--frag-bias-correct Reference/Bos_taurus.UMD3.1.dna.toplevel.fa
02.Align/con/bovine_con_1/accepted_hits.bam
Cuffnorm은 각 샘플의 유전자 발현값을 샘플 간의 비교가 가능하도록 보정을 수행한다. Tophat의 결과 파일인 BAM파일이나 Cuffquant의 결과인 CXB파일이 입력파일로 사용되며, --library-norm-method를 이용하여 보정 방법을 설정할 수 있다. Cuffnorm에서는 FPKM, geometric, quartile방법들을 지원한다.
mkdir -p 06.Cuffnorm
cuffnorm --output-dir 06.Cuffnorm
--labels Control,Experiment
--num-threads 8
--library-type fr-firststrand
--library-norm-method classic-fpkm
--output-format simple-table
05.Cuffquant/con/bovine_con_1/abundances.cxb,
05.Cuffquant/con/bovine_con_2/abundances.cxb,
05.Cuffquant/con/bovine_con_3/abundances.cxb
05.Cuffquant/exp/bovine_exp_1/abundances.cxb,
05.Cuffquant/exp/bovine_exp_2/abundances.cxb,
05.Cuffquant/exp/bovine_exp_3/abundances.cxb
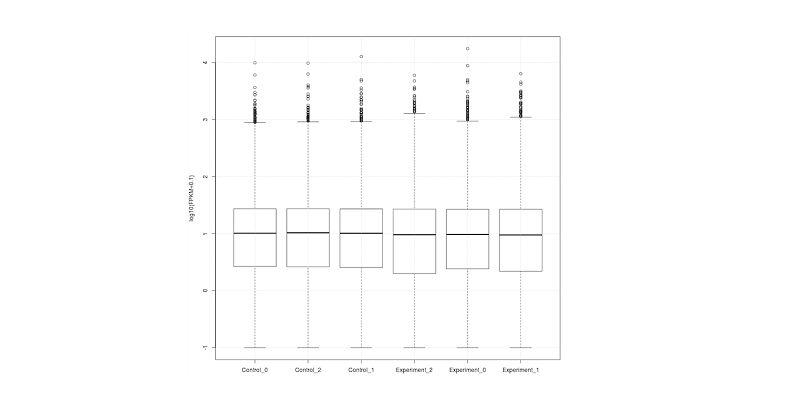
보정된 발현량과 count tables이 결과 파일로 생성되며 그 중 genes.fpkm_table의 데이터를 이용하여 샘플 별 유전자에 대한 발현량 비교가 가능하고 이를 통해 boxplot, scatter plot을 그려서 시각화할 수 있다.
Boxplot은 각 샘플 간 발현 양상을 비교하는데 유용하다. 유전자 레벨에서 샘플 간 비교하기 위해서는 cuffnorm의 결과 파일 중 genes.fpkm_table을 이용해야한다. Boxplot에서 샘플 간 발현 양상이 균등하지 않다면 차등발현유전자 탐색에 좋지 않은 영향을 줄 수 있으므로 다른 보정 방법을 이용해야한다.
# R program
data <- read.table("genes.fpkm_table", header=T, row.names=1)
# box plot
data_sel <- data[which(apply(data,1,max)>0.3),]
png("genes.fpkm_table.boxplot.sel_max.png", width=800, height=800)
boxplot(log(data_sel+0.1,10), xlab="Samples", ylab="log10(FPKM+0.1)")
grid()
dev.off( )
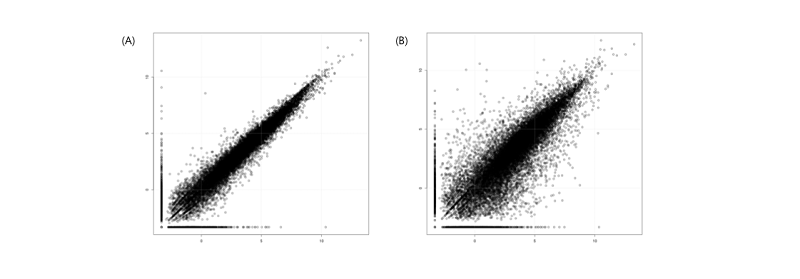
해당 샘플이 대조군 혹은 실험군을 잘 대변하고 있는지 샘플 간의 발현 유사도를 확인 하는 것은 scatter plot과 MDS plot을 통해 가능하다. scatter plot을 통하여 y=x 선을 기준으로 멀리 떨어져 분포되었다면 샘플 간의 발현 양상이 서로 다른 것이며(Figure 9.B), 가깝게 분포될 수록 샘플 간의 발현 양상이 비슷한 것이다(Figure 9.A). 즉, 같은 그룹에 속하는 반복 실험의 샘플일 수록 y=x선에 가까운 분포를 가져야한다. MDS는 PCA와 비슷한 분석으로 모든 샘플 간의 발현 양상을 2차원 혹은 3차원의 요소로 나타내는 방법이다. 같은 그룹이라면 그래프 상에 하나의 군으로 묶여야 데이터의 적합성을 판단할 수 있다.
# R program
data <- read.table("genes.fpkm_table", header=T, row.names=1)
# scatter plot
png("genes.fpkm_table.scatter.C1_vs_E1.png", width=800, height=800)
plot(log(data_sel[,3]+0.1,2), log(data_sel[,5]+0.1,2))
grid( )
dev.off( )
차등발현유전자 탐색
Cuffdiff 과정은 두 그룹의 샘플 별 유전자 발현 정도를 비교하여 차등 발현 유전자를 탐색하는 과정이다. 다양한 비교군으로 샘플을 나누어 분석을 진행할 수 있으며, Sample 간의 비교가 아닌 그룹의 유전자 간의 비교가 가능하다. Cuffquant의 결과 파일을 입력 파일로 사용하여 진행한다.
mkdir -p 07.Cuffdiff
cuffdiff --output-dir 07.Cuffdiff
--labels Control,Experiment
--frag-bias-correct Reference/Bos_taurus.UMD3.1.dna.toplevel.fa
--multi-read-correct
--num-threads 8
--library-type fr-firststrand
--library-norm-method classic-fpkm
--dispersion-method pooled
05.Cuffquant/con/bovine_con_1/abundances.cxb,
05.Cuffquant/con/bovine_con_2/abundances.cxb,
05.Cuffquant/con/bovine_con_3/abundances.cxb
05.Cuffquant/exp/bovine_exp_1/abundances.cxb,
05.Cuffquant/exp/bovine_exp_2/abundances.cxb,
05.Cuffquant/exp/bovine_exp_3/abundances.cxb
Cuffdiff 실행 결과로 유전자, isoforms, CDS, primary transcripts 타입에 대하여 FPKM, counts, read group tracking에 대한 파일과 바이어스 정보 파일 등 총 23개의 결과 파일이 생성된다.
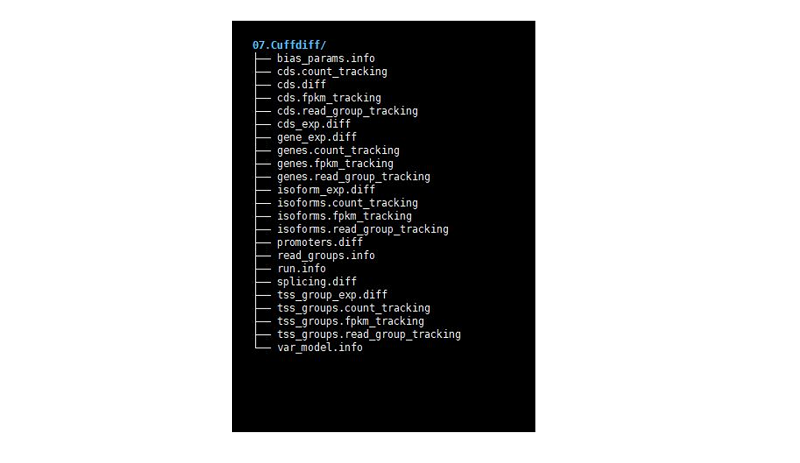
gene_exp.diff 파일에서 grep 명령어를 사용하여 NOTEST를 갖는 데이터를 제거하고 Test ID, 두 그룹의 FPKM값, log2(fold change)값, p value, q value를 추출하여 gene_exp.TEST.diff로 저장한다. R프로그램에서 이 파일을 불러 log2(fold change)에서 Inf, -Inf를 갖는 행을 제거하고 p value 0.05를 기준으로 0.05미만인 데이터들을 저장하여 차등발현유전자를 분류한다.
mkdir 09.Explore_DEG
cd 09.Explore_DEG
cp ../07.Cuffdiff/gene_exp.diff ./
grep -v NOTEST gene_exp.diff | cut -f 1,8,9,10,12,13 > gene_exp.TEST.diff
# R program
data <- read.table("gene_exp.TEST.diff", header=T, row.names=1)
data_sel <- data[which(data[,3]!=Inf),]
data_sel2 <- data_sel[which(data_sel[,3]!=-Inf),]
data_de <- data_sel2[which(data_sel2[,4]<0.05),]
data_node <- data_sel2[which(data_sel2[,4]>=0.05),]
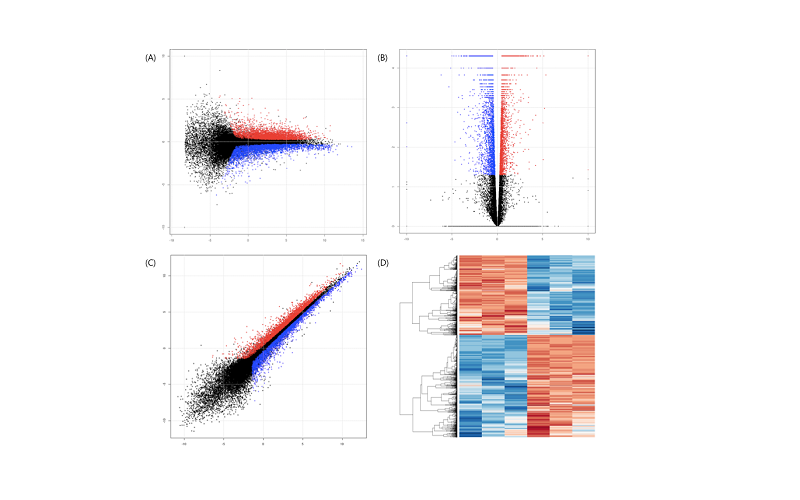
분류된 정보를 사용하여 volcano plot,. scatter plot,. MA plot, Heatmap을 생성할 수 있으며 명령어는 아래와 같다.
# Volcano plot
png("gene_exp.TEST.diff.Volcano.png", width=800, height=800)
plot(data_node[,3], -log(data_node[,4],10),
xlab="log2(foldchange)", ylab="-log(P-value)", cex=0.3, ylim=c(0,5))
points(data_de[,3], -log(data_de[,4],10), col=2, cex=0.3)
grid( )
dev.off( )
# scatter plot
png("gene_exp.TEST.diff.scatter.png", width=800, height=800)
plot(log(data_node[,1],2), log(data_node[,2],2),
xlab="log2(FPKM of Control)", ylab="log2(FPKM of Experiment)", cex=0.3)
points(log(data_de[,1],2), log(data_de[,2],2), col=2, cex=0.3)
grid( )
dev.off( )
# MA plot
png("gene_exp.TEST.diff.MA.png", width=800, height=800)
plot(0.5*(log(data_node[,1],2)+log(data_node[,2],2)),data_node[,3], xlab="M=0.5*(log2(FPKMofCon)+log2(FPKMofExp))",
ylab="A=log2(foldchange)", cex=0.3)
points(0.5*(log(data_de[,1],2)+log(data_de[,2],2)),data_de[,3], col=2, cex=0.3)
grid( )
dev.off( )
# Heatmap
data_heatmap <- data_sel[which(data_sel[,5]<=0.05),c(1,2)]
data_heatmap <- as.matrix(data_heatmap)
png("gene_exp.TEST.diff.Heatmap.png", width=800, height=800)
heatmap(log(data_heatmap+0.1,2), scale="row", cexRow=0.4, cexCol=1.0, margins=c(10,10))
dev.off( )
volcano plot의 x축과 MA plot의 y축은 log2 fold change를 나타낸다. 따라서, 0을 기준으로 up regulation과 down regulation 유전자의 양상을 파악할 수 있다. 또한, volcano plot의 y축은 통계량을 나타내고, MA plot의 x축은 유전자 발현의 정도를 나타낸다. Heatmap은 샘플과 유전자 간의 발현량을 시각화하고, hierarchical clustering을 통해 발현 패턴을 비교할 수 있는 전통적인 방법이다.
Summary
RNA-seq 데이터를 이용한 전사체분석은 전사체 수준에서 생물학적인 현상을 이해하기 위한 도구로 대중화되어가고 있다. 다양한 분석 파이프라인 중 Tuxedo protocol은 생물학적인 현상과 적합한 통계분석을 잘 반영한 분석 파이프라인이다. 본 실습에서는 소(Bos Taurus)의 RNA-seq 예제 데이터를 활용했으며, RNA-seq 데이터의 퀄리티를 향상시키기 위한 전처리단계부터 전사체 분석에 필요한 데이터를 준비하는 과정은 물론, Tuxedo protocol을 이용한 차등발현유전자 탐색까지 설명했다.

