
Background
Next generation sequencing (NGS) is a term used for massively parallel sequencing technology that was developed after the Sanger and the Maxam and Gilbert chemical degradation sequencing method. In NGS technology the desired molecule to be sequenced is broken into small pieces, which are then ligated to adapters for random reading. Since the template is broken into many smaller pieces its read length is generally smaller than the Sanger sequencing method. However some latest methods like single molecule real time (SMRT) and Oxford Nanopore have overcome that drawback. These technologies have longer reads, and thus are more accurate to generate the consensus sequence. Due to its affordability NGS has now become a common tool of use in several fields of biological sciences. NGS generates large amounts of genomic data that can be used to detect genetic variants related to functional alterations. Single Nucleotide polymorphisms (SNPs) are the most abundant type of molecular markers and their high density facilitates interrogation by different genetic approaches. These include large-scale genome association analyses, genetic analysis of simple and complex disease states, genomic predictions and population genetic studies. The use of NGS has enabled to identify SNPs across genomes and allowed the development of pre-designed SNP chips for widespread testing of SNP associations with specific phenotypes of interest.
NGS has led to characterization and quantification of a whole range of “omics” like genomics, transcriptomics and Epigenomics. Omics are essential to understand the mechanisms and functions of different molecules. Different type of NGS sequencing can be used depending upon objective of the project (Figure1). Several livestock genomes have been sequenced recently using NGS. Among livestock species Bos taurus has been the most highly sequenced species followed by Sus scrofa (Table 1). Fast and accurate acquisition of the genome sequence has led to genome-wide identification of causal common and rare variants. Identification of these candidate mutations has enabled the researchers to address phenotypic diversity among livestock species and breeds. Taking advantage of NGS data Sharma et al. (2017) identified 18 mutations involved in Mendelian diseases in Hanwoo cattle of Korea. This information could further be used in a customized SNP chip for this breed. It has also allowed the researchers to study copy number variations (CNVs), genes involved in different pathways and metagenomics. RNA-seq is another popular approach to quantify the expression of genes involved metabolic pathways (Salleh et al., 2017). It has also been used to identify splice variants accurately by mapping sequence fragments onto a reference genome (Suarez-Vega, et al., 2017).
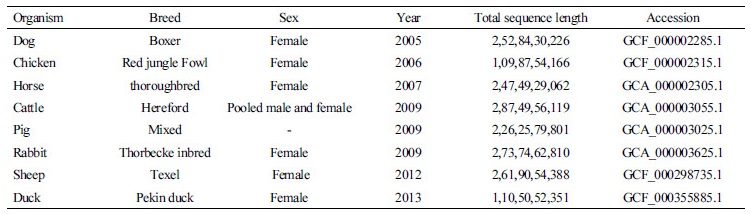
Table 1. Status of next generation sequencing in livestock (www.ncbi.nlm.nih.gov)

|
|
|
#The SRA stores raw sequencing data and alignment information from high-throughput sequencing platforms in NCBI |
|
NGS has also made it possible to study genome-wide epigenetic modifications. Epigenetics may provide information about heritability of complex traits and diseases, imprinting and silencing of transposons which could be of much help in animal breeding (Triantaphyllopoulos et al., 2016). Epigenetic modifications are mediated by small RNAs (sRNAs). Studying methylome allows us to study relationship between sRNA and DNA methylation. NGS based methylome analysis provides a better understanding of methylation patterns across the genome. A better understanding of DNA methylation and other epigenetic modifications will help us establish a relationship among cellular, molecular, physiological and immune responses that play a role in disease resistance.
Studies have demonstrated the value of NGS technologies for molecular characterization, ranging from metagenomic characterization of unknown pathogens or microbial communities to molecular epidemiology and evolution of viral quasispecies (Jose et al., 2017, Yang et al., 2016). Moreover, high-throughput technologies now allow detailed studies of host-pathogen interactions at the level of their genomes (genomics), transcriptomes (transcriptomics), or proteomes (proteomics). The application of high-throughput NGS platforms and their typical low-cost per information content has revolutionized the resolution with which these processes can now be studied. In this paper we review the applications and impact of NGS on livestock species.
Role of NGS in Livestock Diseases and Other Complex Traits
Next generation sequencing of livestock species had allowed a better understanding of their genome, transcriptome and epigenome. Among the livestock species dog was the first livestock animal to be sequenced in the year 2005 (Table 2). Dog has been a loyal companion to humans for thousands of years now. Due to human influence dogs have evolved into several different breeds ranging from difference in size, shape, color and behavior. This human selection had also led to various health issues in these animals. With the use of NGS data researchers have been able to identify key mutations involved in several dog diseases like Lundehund syndrome (LS), a severe gastro-enteropathic disease in the Lundehund dog. NGS pointed towards the association signal on CFA 34 for the LS disease (Metzger et al., 2016). In Golden Retrievers GWAS-guided fine mapping by targeted-NGS has identified novel mutation associated with Generalized progressive retinal atrophy (Downs et al., 2014) and in Tibetian Spaniels/Terriers Downs & Mellersh (2014) identified a short interspersed nuclear element insertion that was associated with Progressive retinal atrophy (PRA). Exome-sequencing has identified the CNGB1 mutation associated with PRA in Papillon and Phalene dog (Ahonen et al., 2013).
Close physical contact of the dogs with the humans puts humans at risk of certain diseases as well. Studying molecular mechanism of the zoonotic transmission from domestic animals to their humans will help address such public health concerns (Oh et al., 2015; Meinel et al., 2014). Comparison of oral microbiomes of dogs and their humans will provide the much needed information about the transmission of any microorganisms which might lead to human diseases.
The data obtained from next generation sequencing has many applications. One amongst them is the identification of the actual expression level of all of the genes that are expressed in essentially any tissue. RNA sequencing (RNA-Seq) allows the quantification of the gene expression for any tissue between two samples. In Horse, Illumina Next Generation Sequencing (NGS) technology was used to identify and characterize the global miRNA expression profile in normal tissues. MiRNAs are important as they provide an insight into various physiological and pathological conditions. Kim et al. (2014) identified a total of 292 known and 329 novel miRNAs in normal horse tissues including skeletal muscle, colon and liver. NGS has also been useful in studying Horse chromosome rearrangement and karyotype evolution (Huang et al., 2014). Diseases like Equine grass sickness and amoebic placentitis were also studied using NGS. NGS provides important opportunities to tackle problems associated with pathogenic illnesses. Based on NGS the identity of the etiological agent for amoebic placentitis in a mare from eastern Australia was confirmed as Acanthamoeba hatchetti (Begg et al., 2014).
NGS has also allowed CNV detection which has opened new avenues for studying genes associated with complex traits in livestock. In Holstein bulls with extremely high and low estimated breeding values (EBVs) for milk protein percentage and fat percentage whole-genome resequencing data identified a total of 14,821 CNVs and 487 differential CNVRs. In addition, 10 genes (INS, IGF2, FOXO3, TH, SCD5, GALNT18, GALNT16, ART3, SNCA and WNT7A), were identified as candidate genes for milk protein and fat traits. In another such study, in Korean Hanwoo cattle a total of 6,811 deleted CNVs were identified using Hiseq 2000 (Illumina, Inc) sequencing data. 33 genes that had high deletion scores were identified to be involved in the domestication process. Their genetic functions were found to be related to nervous transmission, neuron motion and neurogenesis. These genes and the nervous system may be associated with the changes in behavior due to domestication (Shin et. al, 2014).CNVs are known to affect a wide range of phenotypic traits and CNVs in or near segmental duplication regions are difficult to track. However read depth approach based on next-generation sequencing had made it possible to detect such CNVs. Bickhart et al. (2012) used NGS to provide the first individualized cattle CNV and segmental duplication maps and genome-wide gene copy number estimates. A comparative analysis between taurine and indicine cattle breeds was made. It was found that the genes related to pathogen- and parasite-resistance, such as CATHL4 and ULBP17, were highly duplicated in the Nelore cattle relative to the taurine cattle, while genes involved in lipid transport and metabolism, including APOL3 and FABP2, were highly duplicated in the taurine breeds (Beef cattle). These CNV regions harbored genes like BPIFA2A (BSP30A) and WC1, suggesting that some CNVs may be associated with breed-specific differences in adaptation, health and production traits. A similar study was performed in Meishan pigs where segmental duplication (SD) map for pigs was constructed. Genome-wide CNV hotspots were found which were significantly enriched in SD regions, suggesting evolution of CNV hotspots to be affected by ancestral SDs. It was also found that the CNV-related and CNV-unrelated genes undergo a different selective constraint and CNVs may be associated with or affect pig health and production performance under recent selection (Jiang et al., 2014). Such information is of much help in the studies where pig is used as a biomedical model to study human diseases.
In dairy and meat type animals, increase in production and betterment of the quality of the produce is an active area of research. Transcriptomics data can facilitate the functional studies where high and low producing animals can be compared and differentially expressed genes could be identified. Further their metabolic pathways could be identified. All this information could be incorporated in breeding programs. Chen et.al (2015) sequenced and characterized divergent marbling levels in FLW beef cattle. RNA-seq data from the Longissimus dorsi muscle was used to identify the genes that were expressed in low and high marbling animals along with differentially expressed genes.
Recently Bovo et al. (2017) demonstrated the potential of NGS dataset mining for viral metagenomics analysis in livestock. In usual practice the unmapped reads from the sequencing projects are discarded as by-products. But Bovo et al mined these reads in 100 performance tested Italian large white pigs. They assembled these reads for viral metagenomics analysis and were able to identify several viruses of the Parvoviridae family. It was found that the pigs were infected with parvovirus. This study validates the usefulness of NGS for viral metagenomics analysis in livestock. In a similar study Singh et al. (2016) sequenced mitogenome in Indian pig using NGS without designing mitogenome-specific primers.
NGS data in European pig identified three loci that were the elongation of the back and an increased number of vertebrae. The three loci were associated with the NR6A1, PLAG1, and LCORL genes. PLAG1 and LCORL are repeatedly associated with stature in other domestic animals and in humans (Rubin et al., 2012). Choi et al. (2015) carried out genome- resequencing analysis of five pig breeds including Korean native and wild pig and provided a comparative analysis of these breeds. Using NGS data they identified 25.5% novel SNPs and 35,458 non synonymous SNPs in 9904 genes which may contribute to traits of interest. They also identified two genes viz. CLDN1 and TWIST1 that could be associated with economically relevant traits.
Apart from giving insights into the diseases NGS is also used for elucidating breed specific SNPs which could further help in exploring the potential of the breed (Barris et al., 2012; Mengistie et al., 2017, Wang et al., 2017). Outcome of such studies could help design better breeding programs and have a practical benefit in the livestock industry.
Role of Next Generation Sequencing in Animal Breeding
Next generation sequencing has opened up new avenues to explore relationship between genetic and phenotypic diversity with high resolution. Many whole genome sequences of livestock from different breeds and species already exist in the public domain and many new sequencing projects are ongoing. This wealth of data allows us to identify genetic markers spanning the entire genome. In the last five years, large numbers of SNPs have been identified in livestock (particularly bovine, porcine, and ovine species) by performing whole-genome association studies (WGAS). These studies can detect statistical associations between economically important traits and SNP markers, leading to the development of custom marker arrays for genomic selection. Till now genomic prediction depended on SNP arrays but using NGS data provides a clear advantage over SNP arrays as it is not bound by the extent of linkage disequilibrium between SNP markers and the causal mutation as the causal mutation is in the data itself. Use of NGS data for genomic prediction is also believed to yield better results (Figure 2). Advantage of using sequencing data for genomic selection is increased with increase in effective population size and size of the reference population (Druet et al., 2014). Increase in prediction accuracy could be achieved if all the SNPs with causal genes are included in the model equation. Perez-Enciso et al. (2015) found that prediction accuracy increased by 40% when causal genes and SNPs were included in prediction equation. However in dearth of correct biological information the accuracy will drop dramatically which was seen in study reported by Perez-Enciso et al. (2015). Only a 4% increase in accuracy was seen with the whole genome sequencing data over the HD array. Whole genome sequencing (WGS) data can increase accuracy of genomic prediction for low to moderately heritable traits in small populations depending upon QTL density, the size of the reference population and the evaluation method used. The use of WGS data was especially beneficial for multibreed predictions can specifically benefit from the use of WGS data (Iheshiulor et al., 2016).
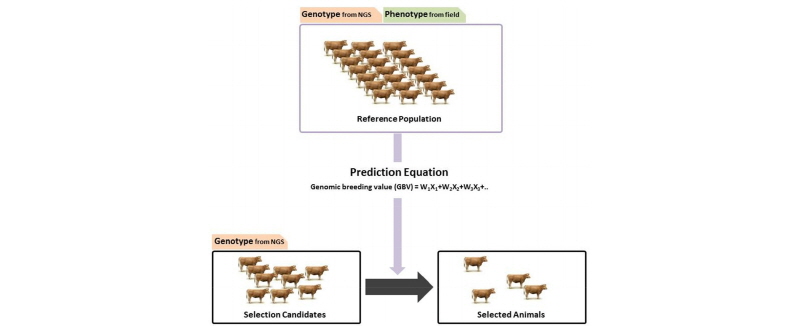
Figure 2.
Genomic prediction in livestock using next generation sequencing (NGS). Reference population has genotype data derived from NGS and phenotype data from the field. Genomic breeding value for each selection candidate is calculated using the prediction equation and best animals are selected for breeding.
Use of NGS in breeding and its practical benefits are yet to be seen. It is an active field of research and its advantages and drawbacks are to be accessed in practical situations before it becomes a standard practice in livestock breeding.
Conclusions
Development in sequencing technologies have opened up plethora of opportunities for researchers to better understand the complex traits and use the information thus gained in livestock breeding programs. NGS has already been used in livestock animals to identify breed specific variants, signatures of selection, causal mutations etc. Current breeding programs mostly rely on marker assisted selection (MAS). And how NGS fairs in the livestock breeding sector still remains to be seen.

