
서론
지난 수십년간 유전능력평가는 개체의 혈통정보 및 표현형기록에 기반하여 진행되어왔으며, Henderson (1975)이 최적선형불편추정법(Best Linear Unbiased Prediction; BLUP)을 제시한 이래로 현재까지 개체의 육종가를 추정하는데 사용되고 있다. BLUP은 개체의 표현형에 영향을 주는 성별, 출생년도 및 출생계절 등 여러 환경요인을 효과적으로 보정하여 개체의 상가적 유전능력을 추정할 수 있는 통계분석 방법으로 추정된 유전능력은 가축의 형질개량을 위한 선발 및 도태의 지표로 사용되고 있다. 그러나 소 등 대가축의 경우 혈통 및 표현형정보를 이용한 유전능력평가는 여러 마리 후대 자손(Progeny)의 도축자료를 확보해야 하며, 이를 위해 5년 이상의 오랜 기간이 소요되는 어려움이 있다(이 등, 2012).
분자유전학 및 유전체학 기법의 발달에 따라 가축의 선발 시 DNA 정보를 선발에 적용하고자 양적형질 좌위(Quantitative Trait Loci; QTL)와 연관된 DNA marker를 이용하는 마커도움선발(Marker Assisted Selection; MAS)이 가능해졌으며 이에 대한 많은 연구가 이루어졌다(Lande and Thompson, 1990; Schulman et al., 1999; Dekkers, 2004). 그러나, MAS 방법은 주요 유전자(Major gene)에 기인한 형질의 경우에는 유용하나 양적형질은 작은 효과를 갖는 다수의 유전자에 의해 영향을 받기에 유전양상을 설명하기에 어려움이 존재한다(조, 2013). 또한 DNA marker의효과를 구명하기 위한 분석 집단의 조성 및 분석에 많은 시간과 비용이 소요되며, 마커정보를 포함한 육종가를 계산하기가 복잡하여 가축 선발에 크게 이용되지 않았다(Hayes and Goddard, 2001; VanRaden et al., 2009).
Meuwissen et al. (2001)은 모의실험을 통해 염색체 내 SNP 표지인자와 QTL간 연관불평형이 존재하는 데이터를 생성한 후 유전체정보와 혈통정보 그리고 표현형정보를 이용하여 개체의 유전체 육종가를 계산하는 유전체 선발(Genomic Selection; GS) 이론을 처음 제안하였으며, 유전체 선발에 대한 이론을 제안한 당시 가축 또는 작물에서 다수의 유전체 정보를 얻기 위해 상당히 많은 비용이 필요했기 때문에 현실화하지 못하고 시뮬레이션을 통해 유전체 선발의 효과에 대해 증명하였다. 그 후, 2002년 인간유전체지도 프로젝트(Human Genome Project; HGP)가 완료됨(Collins et al., 2003)에 따라 인간뿐만 아니라 가축에 있어서도 전체 DNA염기서열이 보고되었다(Eck et al., 2009; Green et al., 2011). 이를 통해 개체간 유전변이가 나타나는 단일염기다형성(Single Nucleotide Polymorphism; SNP) 표지인자를 이용하여 대량의 유전정보를 단시간에 얻을 수 있는 DNA 분석기술이 급속히 발달하였고, 더불어 유전자형 분석비용의 감소를 가져왔다. 이후 GS를 위한 통계적 분석방법에 대한 연구가 활발히 진행되었다(Garrick, 2007; VanRaden, 2007; Hayes et al., 2013). VanRaden et al. (2009)은 기존의 BLUP모형에서 사용하는 혈통기반의 혈연계수행렬 대신 유전체 정보를 이용하여 구성한 혈연계수행렬을 모델에 적용하는 방법을 제안하였다. GBLUP (Genomic Best Linear Unbiased Prediction) 방법은 표현형질과 관련된 유전자가 유전체 전체에 넓게 퍼져 있고 각각의 유전자는 작은 효과를 가진다는 무한소 모형(Infinitesimal Model)을 기반으로 한다(Hayes et al., 2009). 그러나 GBLUP 방법은 오직 유전체 정보를 갖는 개체만 유전능력 평가가 가능하며, 유전체 정보 없이 표현형 정보를 갖는 개체에 대한 동시 분석은 불가하다. Misztal et al. (2009)은 유전체 정보가 없는 개체와 유전체 정보를 갖는 개체간 결합하여 혈연관계를 추정하는 방법(Combined Pedigree and Genomic Relationship Method; H matrix)을 증명하였고, 이를 통해 단일 통계모형에서 두 가지 형태의 데이터를 결합하여 개체의 유전능력을 평가하는 ssGBLUP (Single Step Genomic Best Linear Unbiased Prediction) 방법을 제안하였다. 이러한 유전체 정보를 이용하여 개체의 능력을 추정하는 연구는 표현형 정보가 생성되지 않은 개체에 대하여 기존평가에 비해 보다 높은 정확도로 조기선발이 가능하여 이로 인해 집단의 유전적 개량량의 증대효과가 나타날 수 있다(Aguilar et al., 2010; Christensen and Lund, 2010; Fernando et al., 2014; Lee et al., 2017; Misztal et al., 2020).
우리나라 국가단위 한우개량체계에서도 유전체선발의 이점을 활용하기 위해 2014년부터 농협 한우개량사업소가 보유한 당대‧후대 검정우와 씨수소의 유전체 자료를 분석하여 국가단위 한우의 참조집단을 생성하였다. 이후 당대 검정을 앞둔 송아지의 유전능력을 추정하여 2017년 국내 최초로 유전체정보를 활용해 당대검정우 455두를 선발하였다. 그러나, 송아지 육종가의 절반을 책임진 어미소에 대한 유전능력평가는 대단히 중요함에도 암소에 대한 선발은 주로 혈통과 외모심사로 이루어져 미흡한 실정이다(신, 2008; Kim et al., 2022). 한우 암소검정사업이 2012년부터 전국 농가를 대상으로 수행되고 있으나, 그 사업규모는 전체 대비 미흡한 실정이며, 농가보유의 암송아지 중 번식우(어미소)로 선발하는 방식은 혈통육종가 및 경험적 판단 등으로 농장마다 다르며, 능력검정이 제대로 이루어지지 않고 있다(한 등, 2018).
따라서 본 연구에서는 우리나라의 일반 한우농가 암소 935두에 대해 BLUP, GBLUP 및 ssGBLUP 방법을 이용하여 경제형질 중 도체중, 등심단면적, 등지방두께 및 근내지방도의 유전모수 및 육종가를 추정하고, 추정된 육종가의 정확도를 비교 분석하여 한우 암소의 유전체 선발체계 구축을 위한 일환으로 수행하였다.
재료 및 방법
1. 공시재료
검정집단(test population)은 일반농가에서 사육중인 한우 암소 935두이었으며 이들의 혈액을 채취하였고(혈액의 채취는 지역 동물위생시험소의 협조를 받아 수행하였음), 혈통정보는 확보된 시료의 개체식별번호를 이용하여 한국종축개량협회의 협조로 한우 개체정보조회를 통해 부, 외조부 및 외증조부(KPN)을 추적하여 총 2,176개의 정보를 분석에 이용하였다. 참조집단(Reference population)으로는 2013년부터 2018년까지 전국에서 도축된 한우 중에서 9,849두를 선정하였고, 이들의 도체중(Carcass weight; CW), 등심단면적(Eye Muscle Area; EMA), 등지방두께(Backfat thickness; BFT) 및 근내지방도(Marbling score; MS)에 대한 도축성적은 축산물품질평가원의 협조로 제공받았다. 혈통정보는 한국종축개량협회의 협조로 한우 개체정보조회를 이용하여 혈통 3세대까지 추적하여 총 18,190개의 정보를 분석에 이용하였다.
유전체자료를 확보하기 위해 한우 암소 935두의 경정맥으로부터 채혈한 혈액들을 항응고제(anticoagulant)가 포함된 튜브에 보관하면서 DNA를 추출하였다. Genomic DNA 추출은 Genome Nucleic Acid Purification Kit (MagExtractor, Toyobo CO., LTD. Osaka, Japan)로 이용하였으며, 200 ng/ul 이상의 고농도, 고순도의 DNA를 준비하였다. 이후 약 50,000개의 단일염기다형(SNP)을 갖는 Illumina Bovine SNP50k Bead Chip version 3 (Illumina, SanDiego, Ca, USA)을 이용하여 개체별 대용량 유전자형 분석을 실시하였다. 참조집단인 한우 9,849두의 조직은 축산물품질평가원의 협조로 확보하였으며, genomic DNA를 추출한 후 한우 935두와 동일한 SNP Chip으로 분석하여 유전체정보를 확보하였다.
2. 유전체 정보 품질평가
SNP에 대해 실시하는 품질평가는 Minor allele frequency, Hardy-Weinberg Equilibrium, call rate 등이 있으며, 품질평가에 대한 임계값 설정기준은 샘플집단의 특성을 고려하거나 임의로 결정하는 경향이 있다(Pongpanich et al., 2010). 본 연구의 SNP 자료의 품질 평가는 Plink 1.9 프로그램을 이용하여 Minor allele frequency (MAF) 가 1% 미만, monomorphic SNP, Hardy-Weinberg Equilibrium (HWE) 10-3 미만, Missing proportion (MSP)이 10% 이상인 데이터를 사전에 제거하여 최종 39,254개의 SNP를 이용하였다.
3. 통계분석
3.1 혈통육종가 추정
각 형질에 대한 상가적 유전효과에 대한 유전모수 및 개체의 육종가를 추정하기 위한 혼합선형모형은 다음과 같다.
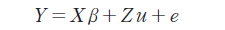
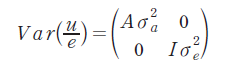
여기서,
Y : 도체형질의 관측치
X : 고정효과(성, 출생년도, 출생계절, 도축년도, 도축계절, 도축장)에 대한 계수행렬
β : 고정효과에 대한 추정치 벡터
Z : 상가적 유전효과의 계수행렬
u : 상가적 유전효과
e : 임의오차의 효과
따라서, E(Y) = Xb, Var(u) = G = Aσa2, Var(e) = R = Iσe2, Cov(μ, e) = 0으로 가정하여 Var(Y) = V = ZGZ′ + R가 된다. 여기서 A는 혈통에 근거한 개체 간의 혈연 관계 행렬(Numerator Relationship Matrix; NRM)이며, I는 대각성분이 1인 단위행렬(Identity matrix), σa2는 상가적 유전분산, σe2는 임의오차 분산이다. 이에 기초하여 혼합모형방정식을 다음과 같이 할 수 있으며(Henderson and Quaas, 1976), 아래의 혼합모형방정식을 BLUPF90 프로그램을 이용하여 도체형질에 대한 개체들의 육종가를 추정하였다.
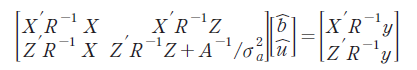
3.2 유전체육종가 추정
(1) 유전체 혈연 행렬
본 연구는 PREGSF90 프로그램(Aguilar et al., 2014)을 이용하여 Allele Frequency Method (GOF) 방법으로 유전체 혈연 행렬(Genomic Relationship Matrix; GRM)을 추정하였다. GOF는 대립유전자의 평균 효과를 0으로 할당하는 방법으로 마커(total number of markers)의 수를 m, 개체의 수(genotyped animals)를 n으로 설정하고, m x n을 M행렬로 표기한다(VanRaden, 2008). M행렬의 원소는 다음과 같이 정의한다.
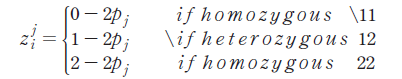
첫 번째 대립인자의 동형접합체가 (11)인 경우는 0으로, 이형접합체 (12)는 1, 두 번째 대립인자의 동형접합체 (22)의 경우 2로 나타낸다. MM′ 행렬은 n x n의 크기로 설정되며, 대각의 원소들은 해당 개체의 동형접합체의 수를 나타내고, 비대각의 원소들은 두 개체들 간에 공통으로 갖는 대립인자의 수가 된다. P는 두 번째 대립인자 빈도(Minor Allele Frequency)를 포함하며 2pj로 표현된다. 관측된 M의 대립유전자 행렬에서 기대 대립유전자형에 대한 값으로 나타나는 P에 대한 행렬의 값을 빼 주게 되면 대립유전자 효과의 평균이 0이 되는(Centered value) Z행렬을 구성할 수 있다. 위에서 계산된 Z행렬을 이용하여 아래와 같은 수식에 의하여 GRM 행렬을 구성할 수 있다.
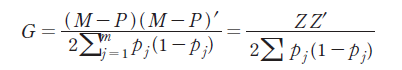
(2) 혈통 및 유전체정보를 결합한 행렬
Legarra et al. (2009)는 유전자형이 존재하는 개체와 유전자형이 존재하지 않는 개체를 동시에 고려하여 결합된 H-matrix를 제안하였으며, Misztal et al. (2009)은 결합된 행렬을 아래와 같이 나타내었다.
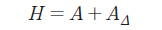
A는 혈통기반의 혈연계수이며, AΔ는 유전체 기반의 혈연계수와 혈통기반의 혈연계수간 차이값에 대한 행렬이며, H는 이들을 결합한 새로운 혈연계수행렬이다. 유전자형이 없는 개체는 첨자 1로, 유전자형이 존재하는 개체는 첨자 2로 나타내면 아래와 같다.
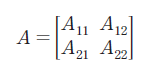
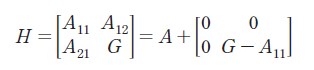
(3) 유전체 유전모수 추정
도체형질에 대한 개체들의 유전체 육종가를 추정하기 위해 다음과 같은 모형을 이용했으며, 혈통과 유전체정보를 혼합한 ssGBLUP 방법 또한 같은 혼합모형을 이용하여 분석하였다.
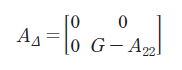
여기서,
Y : 도체형질의 관측치
X : 고정효과에 대한 계수행렬
b : 고정효과에 대한 추정치 벡터
Z : 상가적 유전효과에 대한 계수행렬
μ : 상가적 유전효과
e : 임의오차의 효과
이에 기초하여 유전자형 분석이 완료된 개체들에 대하여 표현형 자료가 있는 개체와 표현형 자료가 없는 개체 모두에 대하여 유전체 육종가를 다음과 같은 혼합모형식을 이용하여 추정하였다.
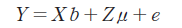
추정된 GRM을 이용하여 G − 1를 추정하였으며, 유전체 육종가를 추정하기 위해 BLUPF90 프로그램을 이용하여 분석하였다. 혈통기반의 혈연정보와 유전체기반의 혈연정보가 결합된 혈연계수 행렬인 H matrix를 이용한 혼합모형식은 다음과 같으며, BLUPF90 프로그램을 이용하여 유전체 육종가를 추정하였다.

3.3 유전력 및 정확도
(1) 유전력(Heritability)
유전력(h2)은 전체분산에서 유전분산이 차지하는 비율로, 다음과 같이 구하였다.
여기서,
σa2 : 상가적 유전분산(additive genetic variance)
σe2 : 환경분산(environmental variance)
(2) 정확도(Accuracy)
혈통육종가와 유전체육종가 그리고 혈통 및 유전체정보를 혼합한 유전체육종가를 추정한 후, 예측된 육종가의 예측오차분산과 추정된 상가적 유전분산을 이용하여 아래의 계산식을 통해 분석하였다.

여기서,
r : 예측된 육종가의 정확도
PEV : 예측된 육종가의 예측오차분산(predicted error variance)
σa2 : 상가적 유전분산
결과 및 고찰
1. 일반분석
1.1 일반성적
한우 암소 935두의 육종가 추정을 위해 2013년부터 2018년까지 도축된 상업축 거세우 9,849두의 도축자료를 이용하였으며, 도체중, 등심단면적, 등지방두께, 근내지방도의 기초통계량은 Table 1과 같다. 도축시 월령은 21개월에서 36개월령으로 분포하며, 평균 31개월로 도체중, 등심단면적, 등지방두께, 근내지방도의 평균 및 표준편차는 각각 441.0±52.0 kg, 95.3±12.0 cm2, 14.3±5.0 mm, 6.1±1.8점(score)으로 분석되었다. 국내의 연구결과를 살펴보면, 원 등(2010)은 2004년부터 2008년까지 도축된 한우 거세우 6,431두의 도체중, 등심단면적, 등지방두께, 근내지방도에 대해 426.8±49.3 kg, 87.9±9.1 cm2, 10.9±4.1 mm, 5.6±2.0점(score)으로 보고하였으며, 구 등(2011)은 2006년부터 2009년까지 도축된 한우 거세우 92,725두의 도체중, 등심단면적, 등지방두께, 근내지방도에 대해 415.23±49.43 kg, 88.29±10.27 cm2, 12.71±5.23 mm, 5.42±1.99점(score)으로 보고하였다. 당 등(2013)은 2007년부터 2011년까지 도축된 한우 거세우 10,441두의 도체중, 등심단면적, 등지방두께, 근내지방도에 대해 423.91±44.90 kg, 91.00±10.11 cm2, 13.63±5.33 mm, 5.60±1.83점(score)으로 나타나 본 연구에서 이용한 도체형질의 표현형 자료가 높은 결과를 보였으며, 도축년도 대비 단순비교해보았을 때, 우리나라의 일반 한우 사육농가 한우의 도체형질에 대한 개량속도는 점차 증가하는 것으로 사료된다.

1.2 정규성 확인
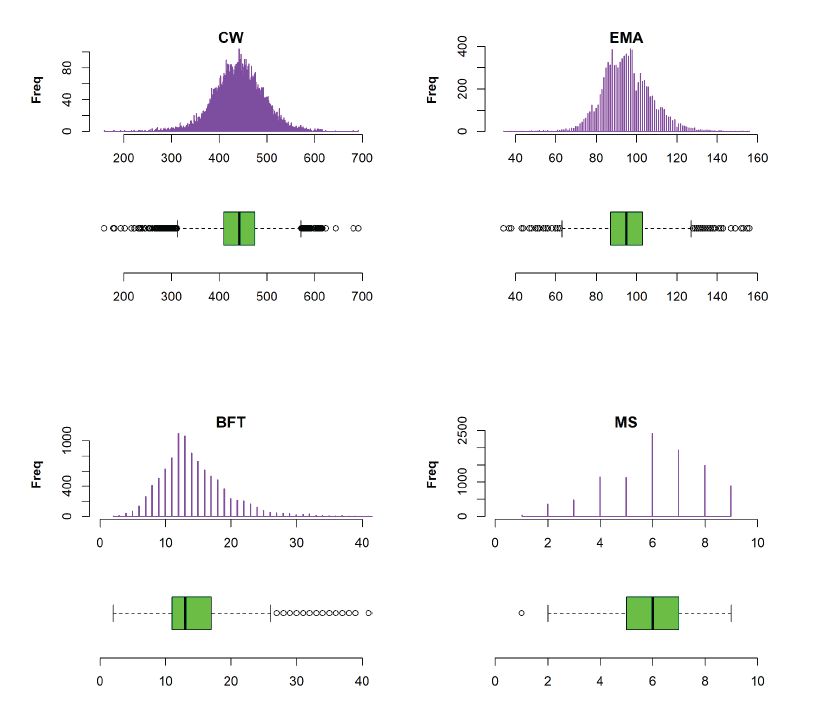
본 연구에서 이용한 표현형 자료의 정규성 확인 결과는 Figure 1과 같다. 도체중, 등심단면적, 등지방두께 및 근내지방도에 대한 집단의 분포와 사분위수-사분위수(Q-Q plot)을 통해 이상치(outlier)를 분석하였으며, 그 결과 각 도체형질은 정규분포의 형태를 볼 수 있었다. 이상치 분석의 결과는 사분위수-사분위수에 벗어난 개체가 있었으나, 이는 유전 및 환경에 의한 형질의 변이 즉, 개체의 다양성을 보여주는 유의한 자료로서 제거하지 않고 분석에 이용하였다.
2. 유전모수 및 육종가 추정
본 연구는 먼저 유전체 정보없이 혈통정보 기반의 혈연계수를 모형에 포함시켜 유전모수를 추정하였을 때, 두번째로 혈통정보 없이 유전체 정보 기반의 혈연계수를 모형에 포함시켜 유전모수를 추정하였을 때, 그리고 세번째로는 혈통정보와 유전체 정보를 혼합한 혈연계수를 모형에 포함시켜 유전모수를 추정하였을 때의 유전분산에 대한 변화를 알아보고자 BLUP, GBLUP 및 ssGBLUP 방법으로 유전모수를 추정하였다.
한우 암소 935두와 참조집단 9,849두에 대한 혈연관계행렬(NRM)을 생성하여 총 20,366개의 혈통정보가 생성되었으며, BLUP을 이용하여 유전력을 추정한 결과, 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.305, 0.324, 0.307 및 0.266으로 나타났다(Table 2). 일반농가 한우의 혈통정보를 이용하여 도체형질에 대한 유전모수 추정한 연구결과를 살펴보면, 원 등(2010)은 한우 거세우 6,431두를 대상으로 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.29, 0.24, 0.35 및 0.36로 나타났다. 당 등(2013)은 한우 거세우 10,441두를 대상으로 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.30, 0.21, 0.42, 및 0.42로 나타나 등지방두께와 근내지방도에서 본 연구보다 높게 나타났다. 국가단위 후대검정집단의 혈통정보를 이용한 도체형질에 대한 유전모수 추정 연구결과는 노 등(2004)이 한우 후대검정우 1,536두를 이용한 결과 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.28, 0.35, 0.39 및 0.51로 나타났다. 황 등(2008)은 한우 후대검정우 2,791두를 대상으로 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.30, 0.37, 0.44 및 0.44로 나타나 등심단면적, 등지방두께 및 근내지방도에서 본 연구보다 높게 나타났다.
|
Table 2. Estimates of variance components and heritability for CW, EMA, BFT, MS using BLUP analysis 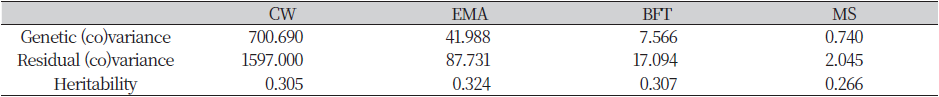
|
|
CW: Carcass weight, EMA: Eye Muscle Area, BFT: Backfat thickness, MS: Marbling score. |
일반농가 한우와 국가단위 후대검정집단 간 유전력이 다른 이유는 크게 각 연구에 사용된 집단의 차이와 크기로 사료되며, 좀더 자세히 살펴보면 첫째, 사육기간에 따른 형질별 표현형의 차이가 있을 것이다. 즉 도축월령이 약 30~32개월인 일반농가 한우와 24개월인 후대검정우 집단간 사육기간의 차이가 표현형에 영향하는 유전적 요인의 비율에 차이로 이해할 수 있을 것이다. 둘째, 사육지역에 따른 환경변이이다. 일반농가 한우의 경우 다양한 환경에서 사육되어지는 반면, 국가단위 집단의 경우 비교적 동일한 사육환경에서 사육되므로 유전모수를 추정할 때 환경 변이에 대한 보정이 좀 더 잘 이루어진 것으로 사료된다. 그리고 분석집단의 크기에 따라 각 표현형질별에 미치는 영향도 어느 정도 반영된 결과로 사료된다.
한우 암소 935두와 참조집단 9,849두 총 10,784두에 대한 유전체 혈연 행렬(GRM)을 생성한 후 GBLUP으로 유전력을 추정한 결과, 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.371, 0.333, 0343, 0.401로 나타났다(Table 3). 이 등(2012)은 거세우 266두를 이용한 결과 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.33, 0.41, 0.40 및 0.50으로 나타났으며, Mehrban et al. (2017)은 한우 거세우 1,183두를 이용한 결과 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.31, 0.43, 0.49 및 0.61로 분석되어 도체중을 제외한 등심단면적, 등지방두께 및 근내지방도에서 본 연구결과 보다 높게 나타났다.
한우 암소 935두와 참조집단 9,849두 총 10,784두에 대해 혈통정보와 유전체 정보를 결합하여 H matrix를 생성하였으며, ssGBLUP으로 유전력을 추정한 결과, 도체형질에서 각각 0.375, 0.336, 0.345, 0.400으로 분석되었다(Table 4). 조(2013)는 유전자형이 있는 후보씨수소 107두와 유전체 정보가 있는 후대검정우 717두 및 유전체 정보가 없는 후대검정우 180두의 총 1,004두에 대해 H matrix를 구축하여 등심단면적, 등지방두께 및 근내지방도에 대해 유전모수를 추정한 결과 각각 0.46, 0.60, 0.63으로 본 연구결과 보다 높게 나타났다.
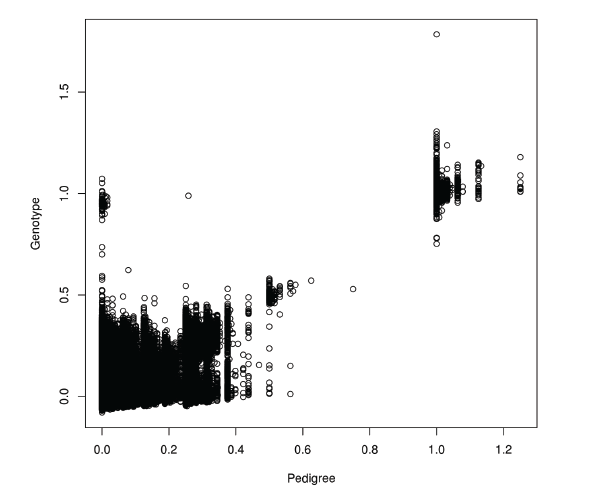
분석방법 간 유전력의 차이를 살펴보면 유전체 정보를 모형에 포함시켜 유전모수를 추정하는 GBLUP과 ssGBLUP의 경우, 혈통정보만 이용한 BLUP에 비해 모든 형질에 대해 유전분산이 증가하고 잔차분산이 감소하여 유전력이 상대적으로 높게 나타났다. 이러한 이유는 혈통정보를 바탕으로 혈연적 유사도의 값을 기대치로 계산하는 혈연관계행렬(NRM)과 유전체정보를 바탕으로 한 유전체 혈연 행렬(GRM)간의 비교를 통해 알 수 있었다(Figure 2). Figure 2의 NRM에서 부와 자손간 혈연관계가 0.5로 나타나는 개체들이 유전체 정보를 이용한 GRM에서는 약 0.4부터 0.6까지 나타나 동일한 혈연을 가진 개체간에도 변이가 존재하였다. 이는 유전체 정보가 있는 개체는 단순히 부와 모의 평균능력을 이용하지 않고 감수분열 단계에서 발생하는 멘델리안 샘플링(mendelian sampling) 효과를 가지기 때문으로 사료된다(Visscher et al., 2006; Hayes and Goddard, 2008; 조, 2013). 그리고 Figure 2에서 크게 두 그룹으로 분리(pedigree 기준 0.6 이하 그룹 및 1.0 내외그룹)되는 것을 볼 수 있는데, 이는 전체적으로 NRM 값과 GRM의 값이 연동되는 것을 알 수 있지만, 이 결과로부터 일부 개체들에서 혈통등록의 오류를 교정하는 효과(예; pedigree 0인 개체들 중 genotype 1 내외인 개체)와 정확한 혈통정보(pedigree 및 genotype에서 1 내외인 개체들)를 확보하여 보다 더 정확한 유전체 육종가를 추정할 수 있는 것으로 사료된다. 반면, GBLUP과 ssGBLUP은 유전력 및 추정육종가에서 큰 차이를 보이지 않았다(Table 5). 이는 ssGBLUP의 H행렬의 혈연계수가 GRM와 동일하게 추정되었기 때문으로 사료된다. H matrix의 혈연계수는 혈통기반의 혈연계수 + 유전체기반의 혈연계수 – 유전자형이 있는 개체에 대한 혈통기반의 혈연계수로 정의할 수 있다. 따라서, 본 연구에서 이용한 집단은 모두 유전체 정보를 가지고 있기 때문에 누락되는 유전체 정보가 없어 결론적으로 유전체 기반의 혈연계수만 이용된 것으로 사료된다.
|
3. Estimates of variance components and heritability for CW, EMA, BFT, MS using GBLUP analysis 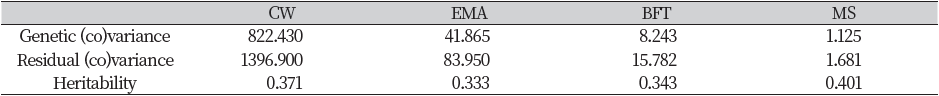
|
|
CW: Carcass weight, EMA: Eye Muscle Area, BFT: Backfat thickness, MS: Marbling score. |
|
Table 4. Estimates of variance components and heritability for CW, EMA, BFT, MS using ssGBLUP analysis 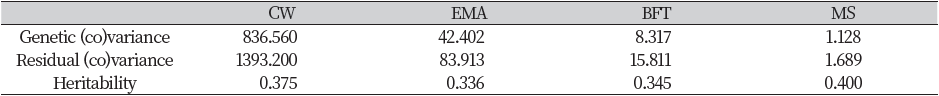
|
|
CW: Carcass weight, EMA: Eye Muscle Area, BFT: Backfat thickness, MS: Marbling score. |
|
Table 5. Correlation of estimated breeding value for carcass traits using BLUP, GBLUP, and ssGBLUP analysis 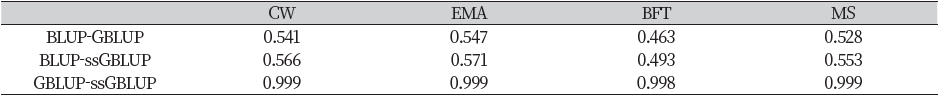
|
|
CW: Carcass weight, EMA: Eye Muscle Area, BFT: Backfat thickness, MS: Marbling score. |
3. 육종가의 정확도 비교
혈통 및 유전체 선발의 핵심은 추정된 육종가의 정확도 일 것이다. 혈통 및 표현형 정보를 이용한 한우암소 935두의 EBV의 정확도는 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.424, 0.430, 0.426 및 0.426으로 분석되었으며(Table 6), 유전체 및 표현형정보를 이용한 GEBV의 정확도는 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.624, 0.613, 0.614 및 0.644로 나타났다. 한편, 혈통, 유전체 및 표현형정보를 이용한 ssGEBV의 정확도는 도체중, 등심단면적, 등지방두께 및 근내지방도에서 각각 0.626, 0.615, 0.616 및 0.645로 분석되었다. BLUP을 이용하여 추정된 육종가의 정확도보다 GBLUP을 이용하여 추정된 유전체 육종가의 정확도가 42.6 ~ 51.2%로 크게 증가함을 볼 수 있었다. 또한 BLUP을 이용하여 추정된 육종가의 정확도보다 ssGBLUP을 이용하여 추정된 육종가의 정확도를 비교하였을 때, 43.02 ~ 51.41%로 크게 증가함을 볼 수 있었다. 그리고 GBLUP보다 ssGBLUP을 이용한 육종가의 정확도가 0.16 ~ 0.33% 증가하였지만 그 차이는 미미하였다. 이는 ssGBLUP 분석 시 유전체기반의 혈연계수 행렬에 모든 유전체 정보가 이용되어 구축되었기 때문이다. 이러한 결과는 한우 암소, 거세우 및 KPN을 대상으로 ssGBLUP으로 유전체육종가를 분석보고한 김 등(2022)의 결과와 비교적 유사하였으며, 암소의 경우에서 BLUP의 육종가 신뢰도 보다 ssGBLUP의 유전체 육종가의 신뢰도가 평균 17~19% 상승되었다. 그리고 한우 거세우를 대상으로 분석보고한 Lee et al. (2017) 및 Lopez et al. (2020)의 연구결과와는 각 형질에 따라 다소의 차이는 나타냈으나 전체적으로 유사하게 ssGBLUP의 결과가 높은 정확도를 보여주었다.

