
Introduction
Due to the generation of highly reliable high throughput sequencing data and the availability of efficient bioinformatics tools, the Next Generation sequencing-based Whole Genome Sequencing (WGS) has become popular among geneticists in the recent past (Park and Kim, 2016). This allows sequencing of the entire genome of a particular individual under reduced cost and time requirements compared with other sequencing technologies, i.e., Sanger sequencing. Further, this facilitates precise genetic characterization and comparative genomic analyses (Parker et al., 2016).
When considering its applications in animals with a particular reference to chickens, many studies have been conducted using WGS targeting different goals, including characterization of genome-wide genetic variants, identification of variants including copy number variants, single nucleotide polymorphisms (SNP), insertion and deletions (INDEL) and selection analysis (Boschiero et al., 2018, Seol et al., 2019, Cho et al., 2022, Gheyas et al., 2022).
The WGS-based studies that recovered consensus sequences for chicken have been performed in some previous studies, i.e., Sohn et al. (2018). However, as many studies highly focused on variant data and specific locations or characteristics, the studies focused on consensus sequences are relatively scarce. This circumstance might be due to two main reasons: (1) either researchers are highly interested in the genomic variations and their effects rather than the sequence, or (2) they have some difficulties in recovering and visualization of the consensus sequence due to a lack of resources (e.g., computational programs, power, space)/ lack of familiarity with such resources. Concerning the second reason, it is efficacious to share the knowledge on appropriate methods/ tools available for obtaining the consensus sequence and visualizing the recovered sequence. Accordingly, this technical report has been written to elaborate on one of such available programs; Geneious Prime® that allows consensus sequence recovery and visualization through both the ‘map to reference’ technique and the ‘de novo assembly’.
In the current report, the consensus sequence recovering through the ‘map to reference’ technique of Geneious Prime® and visualizations of obtained results have been discussed, using the WGS data of one of the Korean native chicken breeds; Ogye, as the raw data set.
Description of the software
Genious Prime® is a software package that includes rich resources of molecular biological tools. It allows for handling, analyzing, and visualizing molecular biological data, including DNA, RNA, and protein. Its ability to obtain well-known external programs as plugins and use them in the same interphase user-friendly manner is considered another importance of this program.
The Geneious Prime 2020 version onwards support Windows (7, 8, 10, 11), and Linux (Ubuntu Desktop LTS, last 2 supported versions) with minimum computational specifications of Intel x86/x86 64 Processor, 2048MB or more memory, 2GB or more free space in hard disk, 1024x768 or higher video resolution. However, it is needless to elucidate that these are ‘minimum’ specifications, and the better the specifications, the better the performance. Further, for handling massive data files, computers with high specifications may required.
When considering the assembly and mapping, Geneious Prime® supports both sanger and Next Generation Sequence (NGS) data for assembly and/or mapping, including Illumina, Ion Torrent, PacBio (CLR/ CCS), Oxford Nanopore and some others. WGS data and the relevant NGS platform can be specified when setting the paired reads. Once the paired reads are set, the reads need to be pre-processed, essentially to remove low-quality sequence reads by trimming and applying some other favorable processing practices, including the removal of duplicate reads and chimeric reads, as preferred. The reads then can be assembled either through the De novo assembly technique or the map to reference technique. Even though the De novo assembly is highly favorable in obtaining an unbiased consensus sequence, requirements to perform De novo assembly, especially the computational resources, including both intensive memory and high run time, are highly challenging (Ye et al., 2012, Zhou et al., 2022). Accordingly, obtaining the consensus sequence by mapping the sequence reads to a reliable and highly related reference sequence is a smart move. Hence, the present paper describes mapping WGS data to a known reference (map to reference technique), and the relevant information has been discussed.
As the resulting consensus sequences are essentially affected by the reference sequence, a highly reliable and suitable reference should be selected. Once the reference sequence (with or without annotations) is ready, the pre-processed reads can be assembled to it. The Geneious prime® provides several mappers for DNA sequence mapping, i.e., Geneious mapper, Bowtie, Bowtie2, and BBMap. By using an appropriate mapper, the mapping process can be conducted. Then based on the settings provided, the consensus sequence can automatically be obtained and visualized.
Data, pre-processing, and mapping
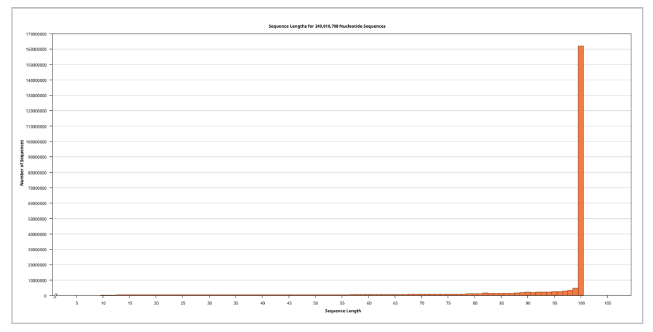
Illumina Miseq ≥ v4 NGS platform-based WGS data for the Ogye Chicken, a Korean Native Chicken, completely black in color, was encoded using Illumina 1.5 with a total of 125,188,143 sequences. The sequence length was 100, and the GC% was 42%.
At first, both forward and reverse raw fastq files were imported to the Geneious Prime® [Geneious Prime 2020.0.1. (https://www.geneious.com), Biomatters, Ltd., Auckland, New Zealand], and paired reads were set (Sequence >> Set paired reads). At this point, the options of pairs of sequence lists and delete unpaired source documents, were selected along with the relative orientation as Forward/ Reverse (inward pointing, e.g., Illumina paired-end). Here, the read technology and expected distance/ insert size, were specified. If the insert size is shorter than twice the read length, usually the paired reads are recommended to be merged.
Then, the reads were trimmed using the BBDuk (BBDuk Adapter/Quality Trimming Version 38.84) plugin of the Geneious Prime® (Annotate and predict >> Trim using BBDuk). Here, the adapters to be trimmed, their desired end of trimming (right-end, left-end), Kmer length, and substitutions, were specified under “Trim adapter”. Further, the trim ends of sequence reads (left-end, right-end, both) and the minimum quality were specified under “Trim low quality”. Minimum overlap and minimum length were also stipulated under the “Trim adapters based on paired overhangs” and “Discard short reads” menus, respectively. These specifications can be changed with the different sequencing platforms used with various adapters, varying complexities of the region of interest, the objective of trimming, and the expected quality of the final result. However, for the current work, default settings were used.
Once the trimming was completed the trimmed reads were normalized for coverage using BBNorm 38.84 version to reduce the computational time required for mapping without affecting the resultant assembly. Then the normalized reads were mapped to the reference. The genome assembly of GRCg7b – chicken whole genome short gun sequence assembly (GCF_016699485.2) of NCBI was used as the reference.
To facilitate an easy and understandable description, it has explained the process of mapping to a randomly selected only one chromosome of the particular assembly; chromosome 16 (NCBI accession number NC_052547.1 [RefSeq sequence]). The complete reference sequence with reference features can be obtained by importing the downloaded GenBank format of NC_052547.1 from NCBI to Geneious Prime® or directly searching the NCBI accession number NC_052547.1 in Geneious prime’s “NCBI” tab. The same process can be practiced for all the other chromosomes as well, to get their consensuses.
Accordingly, the trimmed reads mapped to the NC_052547.1 reference sequence by reaching the Align/Assemble >> Map to reference. Here the pre-imported reference sequence (single sequence) was chosen, under the data tab. In the method tab, different options for mappers are available. Among them, the Geneious mapper was used for this study. The desired sensitivity was set to Medium-Low sensitivity/Fast. To obtain the structural variants, the option of “Find structural variants, short insertions and deletions of any size” was marked. In the “Trim Before Mapping” tab, “Do not trim” is selected as the reads were trimmed earlier with the BBDuk trimmer.
When customizing the “Results” preferences regarding the consensus sequence, “Save consensus sequence” was enabled. In the “Consensus Options” menu, the recommended “Highest Quality 60%” was selected, and the settings were modified to call the reference bases when there is no coverage and when the coverage is less than five. Further, the program was set to “trim to reference sequence” and “Ignore reads mapped to multiple locations”. Moreover, the sanger heterozygotes were called. In the “Advanced” tab, the minimum mapping quality, which determines the minimum confidence of the accurate mapping, was set to 20 (99% confidence).
After completing the assembly, the assembly report was analyzed, and the consensus sequence was also checked. The contigs were visualized to check the mapped regions, gaps, variations, and consensus sequence.
Unlike in many other assembly pipelines, the Geneious Prime® clearly visualizes the resultant assembly (Contig) in a highly understandable manner, importantly with customizable settings and zooming options. These features are utterly valuable in elaborating assembly results, especially with proper coverage, structural variants, and gap regions.
Results
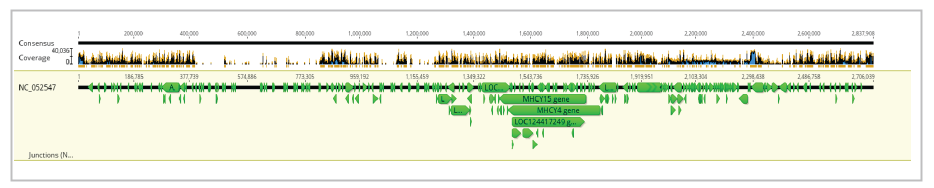
The assembly of Ogye NGS sequences by mapping to NC_052547.1 resulted in a whole assembly (even outside the reference), with a length of 2,837,908 bp within 4 hours and 15 minutes. For mapping 3,325,858 sequences have been utilized, and the pairwise identity between the current assembly and the reference was 76.6%. Frequencies of adenine (A), thymine (T), guanine (G), cytosine (C), and GC contents are 24.8%, 24.4%, 26.2%, 24.6%, and 50.6 percent, respectively. The length of the reference sequence was 2,706,039 bp, and the resulted consensus was 2,653,414 bp.
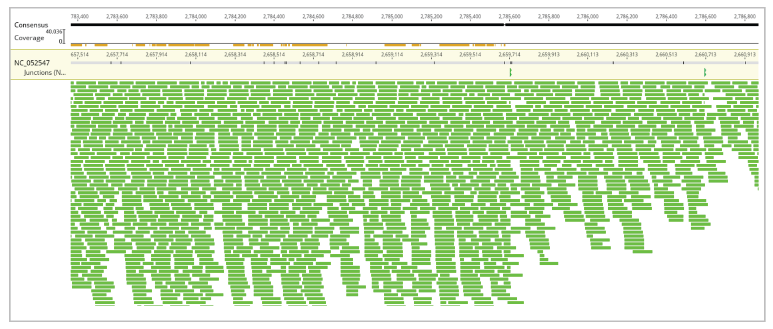
Visualization of the whole assembly (for chromosome 16), manner of sequence assembly (zoomed), and assembly zoomed as the bases of reads, reference and consensus sequence are clearly visible, have been presented in Figures 5, 6, and 7, respectively.

Discussion
Geneious mapper is fast, highly sensitive, supportive for soft trimmed reads, and contains iterative modes. As it is capable of calling for structural variants, including SNPs and INDELs. It is worth searching for polymorphisms in experimental data against the reference data. Besides, it is highly accurate, and it can be handled effortlessly even under default settings without affecting the proper performance of the mapping process. Moreover, it provides iterative facilities for a more precise mapping result (Kearse et al., 2012).
This mapper has been used successfully in various scientific studies including Hinckley et al. (2020), Lee et al. (2021), Huang et al. (2021), and Papp et al. (2022).
In the GRCg7b assembly, there are 39 autosomes and 2 sex chromosomes. Among them, only chromosome 16 was selected and hence a giant majority is not mapped here. Further, the consensus sequence generated contains some ambiguities denoted with “N” s. Sometimes, this is due to the occurrence of such ambiguities (“N” s) in the reference sequence. Accordingly, high dependency on the reference can be considered one of the major limitations of this technique that can affect the accuracy of the resulting consensus sequence.Conclusion
Applying the explained technical approach of “map to reference” with Geneious Prime® for WGS data can effectively assemble any genome and obtain their consensus sequences with considerably lesser computational requirements and run time than the De novo assembly. The obtained consensus sequences of genomes can then be annotated and used for various molecular genetics approaches.

