1Department of Animal Science, Chungnam National University, Daejeon, 34134, Republic of Korea
2Institute of Agricultural Science, Chungnam National University, Daejeon, 34134, Republic of Korea
3Department of Bio-AI Convergence, Chungnam National University, Daejeon, 34134, Republic of Korea
4Division of Animal & Dairy Science, Chungnam National University, Daejeon, 34134, Republic of Korea
Correspondence to Seung Hwan Lee, E-mail: slee46@cnu.ac.kr
Volume 9, Number 2, Pages 57-77, June 2025.
Journal of Animal Breeding and Genomics 2025, 9(2), 57-77. https://doi.org/10.12972/jabng.2025.9.2.2
Received on May 16, 2025, Revised on June 24, 2025, Accepted on June 24, 2025, Published on June 30, 2025.
Copyright © 2025 Korean Society of Animal Breeding and Genetics.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0).

QMSim is a powerful simulation program designed to emulate large-scale genomic data and complex population structures, making it a valuable tool for developing efficient animal breeding program. This protocol provides a step-by-step guide for installing and running QMSim, along with detailed explanations of essential commands and parameters used in the input files. By enables the implementation of diverse simulation scenarios and facilitates the validation and evaluation of proposed genomic selection strategies, thereby contributing to the development of optimized breeding programs.
QMSim, Simulation, Genomic Selection, Genetic Evaluation
지속적인 인구 증가와 함께 고품질 축산물에 대한 수요가 증가함에 따라, 효율적인 동물 육종 기술은 식량안보 확보와 축산업의 경제적 성장에 핵심적인 역할을 하고 있다(Thornton. 2010; FAO., 2020). 최근 동물 육종 분야는 유전체 정보 활용과 빅데이터 기반 정밀축산 기술 등의 발전을 통해 대량의 유전체 및 표현형 데이터를 통합 분석하여 육종 효율성을 극대화하는 방향으로 발전하였다(Meuwissen et al., 2016; Morota et al., 2018; Stock et al., 2013; VanRaden. 2020; Nayeri et al., 2019). 유전체 데이터는 동물의 표현형을 정밀하게 예측할 수 있는 잠재력을 지니고 있다. 그러나 집단 크기, 마커 밀도, 환경 요인 등 복합적인 영향으로 인하여 분석 과정이 복잡하며, 많은 비용과 시간이 요구된다(ZHU et al., 2017; Hall., 2016; Wanjala et al., 2023; Santos et al., 2017).
이러한 경우 다양한 요인을 통제하면서 단기간에 개량 과정을 모의 실험할 수 있는 시뮬레이션이 효과적인 방법으로 활용될 수 있다(Medrano et al., 2010; Simianer et al., 2021; Scheper et al., 2016). 시뮬레이션 실험을 통해 번식전략을 수립하고 유전적 개량량을 최적화하며, 가축의 예상 성과를 예측할 수 있다(Lopes et al., 2019; Andonov et al., 2017). 현재 가축 육종에서 활용할 수 있는 시뮬레이션 프로그램으로는 AlphaSim, ZPLAN+, SelAction, QMSIM 등이 있다. AlphaSim의 경우 단일염기다형성(SNP) 및 양적형질핵산(QTN) 시뮬레이션이 가능하여 정밀한 표현형 예측을 지원하며(Faux Am et al., 2016), ZPLAN+는 특정 번식 전략의 장기적 영향을 예측하여 육종 전략을 최적화 하는데 도움을 준다(Täubert et al., 2010). 또한, SelAction은 실제 육종 프로그램에서의 선발반응과 근친교배율을 예측하는 기능을 갖추고 있어 실용적인 육종 전략 수립에 유용하게 활용된다.(Rutten et al., 2002).
특히, QMSim은 대규모 유전체와 복잡한 계보 구조를 모방하여 가축 개체군을 시뮬레이션 할 수 있는 프로그램으로, 다양한 개체군 구조를 모델링하고 양적 형질을 시뮬레이션 하여 육종전략 평가, 유전적 개량량 예측, 근친교배의 장기적 영향 등을 분석하는데 활용될 수 있다(Sargolzaei et al., 2009; Scheper et al., 2016; Sargolzaei et al., 2009) (Table 1). 이 프로그램은 젖소, 돼지, 육우, 가금류, 수산 양식 종 등 여러 가축종에서 유전체 선발과 육종 효율성 향상을 위한 중요한 도구로 활용되어 왔다(Seno et al., 2014; Yin et al., 2024; Plotzki Reis et al., 2019; QiXin et al., 2016; Wang et al., 2019; Khalilisamani et al., 2022).
Table 1. Main features, strengths, and limitations of the simulation programs are summarized.
| 프로그램 | 주요 기능 | 장점과 단점 |
|---|---|---|
| AlphaSim | 유전체 선발 및 변이 프로그램 시뮬레이션 |
• 다양한 유전적 변수 (SNP, QTN 등) 조절 가능 • 유전체 선발 및 유전자 편집 지원 |
| ZPLAN+ | 다양한 육종 전략 최적화 및 육종 프로그램의 경제적 평가 |
• 대규모 데이터 시뮬레이션에는 적합하지 않음 • 경제적 수익-비용 분석 지원 • 근친교배 방지 및 최적 선발전략 제공 |
| SelAction | 선발 반응 및 근친교배율을 예측하여 다양한 육종전략 수립 |
• 복잡한 설정과 유전체 시뮬레이션 기능 미흡 • 빠른 연산 속도 • 대규모 유전체 데이터 처리 미흡 |
| QMSim | 대규모 유전체 데이터 시뮬레이션 |
• 대규모 유전체 시뮬레이션 지원 (마커의 경우 최대 40만) • 다세대 계통 및 복잡한 개체군 구조 구성, 선발방법, 근친교배 최적화, 염색체 구성, 유전적 변이 및 오류 등 다양한 요소를 세밀하게 반영하여 분석 가능 • 복잡한 설정 파일 |
본 논문은 QMSim 프로그램의 설치 및 실행 절차와 함께, 시뮬레이션 설정을 위한 매개변수 파일 내 주요 명령어들의 기능과 사용법을 체계적으로 설명하였다.
이 프로그램은 시뮬레이션에 필요한 다양한 매개변수를 지정하는 매개변수 파일을 필요로 하며, 이 파일은 다섯개의 주요 섹션(GLOBAL parameters, Historical population, Populations, Genome, Output options)으로 구성되어 있다(Figure 1).

Figure 1. Basic structure of the parameter file used in QMSim
각 섹션 내에서 명령어의 순서는 중요하지 않지만, 모든 명령어는 반드시 세미콜론(;)으로 끝나야 하며, 세미콜론이 누락될 경우 오류 메시지가 출력되고 프로그램이 종료된다. 또한, 필수적으로 사용되지 않는 명령어의 설정은 사용자의 선택에 따라 추가하거나 삭제할 수 있다.
Global parameter section은 전체 시뮬레이션 과정에서 적용되는 매개변수들을 설정하는 구간이다(Figure 2). 다음은 주요 명령어에 대한 설명이다.

Figure 2. Global parameter section 명령어 예시
title = “title_name”; 을 사용하여 시뮬레이션의 제목을 지정할 수 있다. 시뮬레이션을 실행하면, seed 파일이 자동으로 생성되게 되는데, 이후 seed = “”; 명령어를 사용하여 해당 seed 파일을 지정하면 이전에 수행한 시뮬레이션을 동일한 조건으로 다시 실행할 수 있다. seed =“”; 명령어를 별도로 지정하지 않아도 시스템에서 자동으로 난수를 생성하여 시뮬레이션을 진행한다.
nrep = num; 명령어를 사용하여 시뮬레이션 반복 횟수를 1부터 20,000번까지 설정할 수 있다. h2 = num; 에는 시뮬레이션 할 형질의 총 유전력(Polygenic +QTL)을 지정하며, 0 ~ 1사이의 값을 설정해야 한다. qtlh2 = num; 은 QTL의 유전력만을 지정하는 명령어로, h2 =num; 과 마찬가지로 0 ~ 1사이의 값을 설정할 수 있다. 만약 qtlh2 = 0; 으로 설정하면, 다유전자(polygenic)효과만을 포함한 시뮬레이션이 실행된다.
phvar = num; 는 형질의 표현형 분산을 설정하는 항목이며, 값의 범위는 0 ~ 10,000,000 까지 지정할 수 있다. no_male_rec; 명령어를 사용하면 수컷 개체의 기록을 생성하지 않으며, 우유 생산량과 같은 성 제한 형질을 시뮬레이션 할 때 활용할 수 있다. 수컷의 표현형 데이터가 없을 때, 표현형 기반 선발과 도태를 적용하면, 수컷은 무작위로 선택된다. joint_pop; 명령어를 설정하면 둘 이상의 개체군을 시뮬레이션 할 때, 하나의 집단으로 통합하여 분석할 수 있으며, 이 옵션을 사용하지 않으면 개체군은 개별적으로 분석된다.
skip_inbreeding; 명령어는 근친교배의 계산을 생략하는 기능으로, (1) 다유전자 효과를 고려하지 않고 모든 유전적 분산이 QTL 에 의해 설명되는 경우, (2) BLUP(최적 선형 불편 예측) 기반 육종가 추정을 하지 않는 경우, (3) 교배계획을 최적화 하지 않는 경우에 사용해도 무방하다. 하지만 이 옵션을 사용할 경우 근친 교배가 가축 개량에 미치는 영향을 고려하지 않기 때문에 결과 해석 시 주의가 필요하다.
Historical population section은 초기 연관불균형(Linkage disequilibrium, LD) 생성과 돌연변이-유전적 부동(mutation-drift) 평형을 확립하기 위한 개체군을 설정하는 구간이다. 동일한 수의 수컷과 암컷 개체로 개체군을 구성하며, 자손은 수컷과 암컷의 생식세포가 무작위로 결합하여 생성된다. 또한 시뮬레이션을 통해 개체군의 크기를 확장하거나 축소 할 수 있다(Figure 3). 다음은 주요 명령어에 대한 설명이다.
begin_hp; & end_hp; 명령어를 사용하여 historical population 매개변수의 시작과 종료를 정의해야 한다. 모든 historical population의 매개변수는 begin_hp; 로 시작하여 end_hp; 로 끝나야 한다.
hg_size = v1 [v2]; 명령어는 historical population 세대 크기를 설정하는 항목으로, v1은 개체군 크기, v2는 해당 크기로 유지할 세대 수를 의미한다. 개체군 크기는 2 ~ 100,000, 세대 수는 0 ~ 150,000 범위 내에서 설정 가능하며, 세대수는 0부터 시작해야 한다. 추가적인 매개변수를 통해 개체군의 확장이나 축소 시나리오를 유연하게 구성할 수 있으며, 구체적인 사용법은 제공된 예제를 참고하면 이해하기가 쉽다(Figure 28).
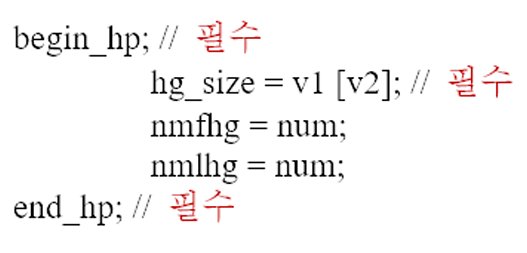
Figure 3. Example command for generating a historical population.
nmfhg 와 nmlhg 는 historical population 에서 수컷 개체의 수를 설정하는 명령어이다. nmfhg = num; 는 첫 번째 historical 세대에서의 수컷 개체 수, nmlhg = num; 는 마지막 historical 세대에서의 수컷 개체 수를 의미하며, 이들을 통해 세대 간 성비 변화를 구현할 수 있다. 각각 1 ~ 100,000의 값으로 설정 가능하며, 해당 명령어가 생략될 경우 전체 개체군은 암수 성비가 동일한 상태로 유지된다.
Population Section은 recent population을 설정하여 시뮬레이션에 사용할 개체군을 생성하는 구간이다. 하나 이상의 개체군을 생성할 수 있으며, founder는 historical population 또는 recent population 내에서 선택할 수 있다. 두 개 이상의 개체군을 분석할 경우 GLOBAL PARAMETER SECTION에서 joint_pop; 명령어를 사용하여 하나의 계보로 통합 분석하거나, 개별적으로 분석할 수 있다. 이 구간을 통해 근친교배율과 추정육종가(EBV)를 평가할 수 있으며, 최대 1000개의 recent population을 시뮬레이션 할 수 있다.
begin_pop = “string”; & end_pop; 명령어는 각각 recent population 매개변수 블록의 시작과 종료를 정의하는데 사용된다. begin_ pop = “string”; 명령어 string 에는 개체군 이름을 지정하며, 하나 이상의 개체군을 생성할 경우, 반드시 각 개체군 마다 begin_pop = “string”; 으로 시작하고 end_pop; 으로 종료되어야 한다(Figure 4).

Figure 4. An example of how to generate two recent populations in the Population section using the begin_ pop and end_pop commands.
두 개 이상의 개체군을 생성할 경우, 첫 번째 recent population의 founder는 반드시 Historical population의 마지막 세대에서 선발해야 한다. 이후 정의되는 개체군의 경우, 앞서 정의된 recent population 중 최대 10개 이전 개체군을 founder로 활용할 수 있으며, QMSim은 입력 파일에 정의된 순서대로 각 개체군을 처리한다.
begin_founder; & end_founder; 명령어를 사용하여 recent population의 founder를 정의한다. male[]; 및 female[]; 명령어를 사용하여 수컷과 암컷의 개체 수(n), 선발 할 집단(pop), 선발 세대(gen), 선발 기준(select) 등을 지정할 수 있다(Figure 5).

Figure 5. Example of how to use commands for setting up founders
선발할 개체의 수(n = num)는 1 ~ 50,000 범위 내에서 지정할 수 있으며, 선발할 개체군(pop = “”)은 “hp”(historical population) 혹은 “string”(다른 개체군 이름)을 입력할 수 있다. 또한, 해당 개체군에서 선발 세대(gen = num)를 0 ~ 2,000 범위 내에 설정할 수 있다. 다만, gen은 선택 항목으로 입력하지 않아도 된다.
선발 기준(select = value option)은 rnd(무작위), phen(표현형기반), tbv(실제육종가치), ebv(추정육종가치)중 선택 가능하며, 기본값은 rnd(무작위)이다. 추가적으로 /l 옵션을 사용하면 낮은 값을 기준으로, /h 옵션을 사용하면 높은 값을 기준으로 개체를 선발한다. 단, select = rnd일 경우에는 /l 및 /h 옵션을 사용할 수 없다. 또한, 가장 먼저 설정되는 수컷과 암컷 founder는 반드시 historical population (pop = “hp”)에서 선발되어야 한다.
recent population의 founder를 설정한 후, 개체군의 구조를 정의해야 한다(Figure 6).

Figure 6. Example of command structure for defining the recent population after founder setup
ls = value [p]; 명령어는 산자수(litter size), 즉 한 개체당 후손(progeny)의 수를 설정하는 항목이다. 두 가지 방식으로 사용이 가능하며, ls = value; 형식으로 단일 산자수 값을 0 ~ 1,000 범위 내에서 지정할 수 있고, ls = value [p]; 형식으로 산자수 [산자수의 확률]을 0 ~ 1 범위 내에서 지정이 가능하다. 산자수는 유전적으로 조절되지 않으며, 입력된 확률을 기반으로 하여 개체군이 샘플링된다.
ng = value; 명령어는 현재 개체군의 세대 수를 설정하는 항목으로, 0 ~ 2,000 범위 내에서 지정이 가능하다. pmp = value; 명령어는 자손 중 수컷 자손의 비율을 설정하는 항목으로, 0 ~ 1 사이의 값을 지정 가능하며, 기본값은 0.5 이다. pmp = value /fix; 옵션을 지정하게 되면, 전체 개체군에서 수컷 비율이 설정된 값과 동일하게 일치하도록 시뮬레이션 된다.
md = value option; 명령어는 교배 설계를 설정하는 항목으로, value 에는 rnd, rnd_ug, minf, maxf, p_assort, n_assort 중에서 선택할 수 있으며, 기본값은 rnd(무작위 교배)이다. rnd_ug 는 무작위 생식세포 결합을 의미하며, 수컷과 암컷의 생식세포 풀에서 두 생식세포를 무작위로 결합하여 후손을 생성한다. 이 방식에서는 한 개체가 여러 개체의 배우자와 교배할 수 있으며, 산자수(ls) 값이 어미당 평균 자손 수로 처리된다. p_assort 및 n_assort 는 유사성 및 비유사성을 기준으로 교배를 설계하는 방식이며, 유사성 판단 기준은 option에 /phen(표현형기반), /tbv(실제육종가 기반), /ebv(추정육종가 기반)를 기준으로 설정할 수 있다. minf 와 maxf는 각각 근친교배율을 최소화하거나 최대화하는 교배 설계를 의미하며, 해당 명령어를 사용할 시 option은 별도로 설정하지 않는다.
sr = v1 [v2]; 및 dr = v1 [v2]; 는 각각 씨수컷과 씨암컷 개체의 교체 비율을 설정하는 항목이다. 기본적으로 모든 세대에서 일정한 비율의 씨수컷과 씨암컷이 도태되며, 추가적인 매개변수를 통해 특정 세대에서의 교체 비율을 조정할 수 있다. v1의 경우 도태될 종축의 비율을 0 ~ 1 범위 내에서 조정하고, [v2]의 경우 교체를 시작할 세대를 지정할 수 있다. 만약 도태 비율보다 개체군 감소율이 높아 개체수가 부족해지는 경우, 시스템이 자동으로 교체율을 조정하며, 개체 수 증가율이 너무 낮을 경우 경고 메시지가 출력된다. v1만 단독으로 설정하여 도태 비율만 별도로 지정하는 것도 가능하다.
sd = value option; 개체를 선발하는 방법을 지정하는 명령어이다. value 에는 rnd(무작위 선발), phen(표현형 선발), tbv(실제육종가 선발), ebv(추정육종가 선발)중 하나를 선택하여 지정할 수 있으며, option으로 /l 을 지정하면 낮은 값(낮은 표현형, 낮은 육종가)을 가진 개체를, /h 를 사용하면 높은 값(높은 표현형, 높은 육종가)를 가진 개체를 선발한다. 기본값은 /h 이며, 선발방법이 rnd(무작위)일 경우 해당 option을 사용할 수 없다.
cd = value option; 은 개체를 도태하는 방법을 지정하는 명령어이다. value 에는 rnd(무작위 도태), phen(표현형 기준), tbv(실제육종가 기준), ebv(추정육종가 기준), age(연령 기준) 중 하나를 선택할 수 있다. 기본값은 age(연령 기준)이며 , option 에 /l 을 사용하면 낮은 값을 가진 개체를 우선 도태, /h 옵션을 사용하면 높은 값을 가진 개체를 우선 도태한다. 하지만. 도태방법(value)이 rnd(무작위) 또는 age(연령기반) 일 경우 option 은 사용할 수 없다.
ebv_est = blup; 명령어는 개체의 육종가(Breeding Value) 추정을 설정하는 항목으로, 사용자가 해당 명령어를 지정해야만 개체의 육종가치를 계산한다. 육종가는 개체가 부모로서 후손에게 전달할 수 있는 유전적 가치를 의미하며, Best Linear Unbiased Prediction (BLUP) 을 이용해 추정된다.
Population section 에서는 생성된 개체군의 출력 데이터를 설정하는 필수적인 명령어가 존재한다(Figure 7).

Figure 7. Example of command settings for defining output data after population setup
begin_popoutput; & end_popoutput; 은 개체군의 출력옵션을 설정하는 필수적인 명령어로, 특정 개체군의 데이터 저장방식을 정의한다.
data; 명령어를 사용하면 유전자형(Genotype) 정보를 제외한 개체 별 정보를 저장할 수 있다. option 에 /gen 세대수1 세대수2 세대수3 … 명령어를 추가하여 특정 세대의 데이터를 저장할 수 있다. 파일은 “개체군 이름”_data_“반복 번호”.txt 형식으로 저장되며, 각 개체의 유전적 요소와 표현형 정보가 포함된다(Figure 8, Table 2).
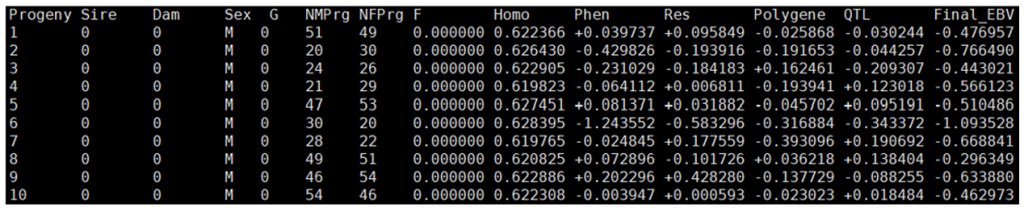
Figure 8. Examples of files generated by the ‘data;’ command.
Table 2. Description of the items contained in the files generated by the ‘data;’ command
| Progeny 개체ID |
Sire 부ID |
Dam 모ID |
Sex 성별 |
G 세대 |
NMPrg 모계 자손 |
NFPrg 부계 자손 |
| F 근교계수 |
Homo 동형접합도 |
Phen 표현형 |
Res 잔차 |
Polygene 다유전자효과 |
QTL QTL 효과 |
EBV 추정육종가치 |
stat; 명령어는 시뮬레이션 된 데이터의 통계 요약을 저장하는 기능을 한다. 해당 명령어를 사용하면 파일이 “개체군 이름”_stat_ “반복 번호”.txt 형식으로 저장되며, 근친교배율, 동형접합도, 표현형 및 개체군 구조 정보의 평균 및 표준편차, 그리고 시뮬레이션 된 개체군의 구조정보가 포함된다(Figure 9).
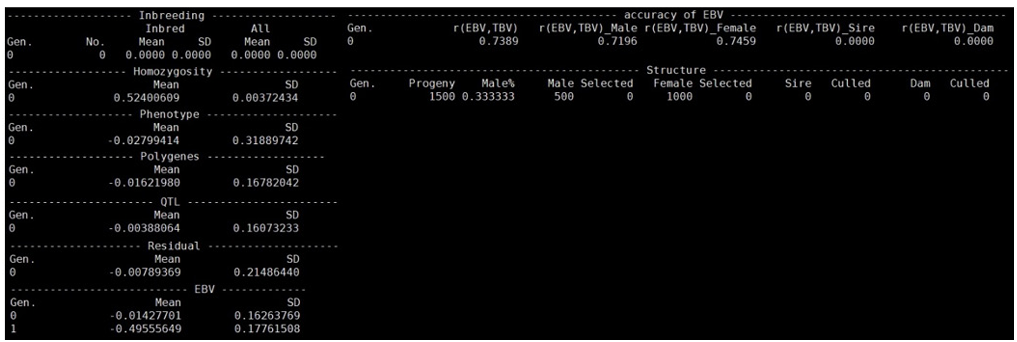
Figure 9. Example of a data file generated by the ‘stat;’ command
genotype; 명령어는 개체의 유전자형 데이터를 저장하는 기능을 하며, 다양한 형식으로 유전자 정보를 출력할 수 있다. option 에는 /binary, /snp_code, /seq, /gen … 를 입력할 수 있다. /binary 는 유전자 데이터를 이진(binary) 형식으로 저장하는 방식이며, /snp_code 는 SNP를 유전자 코드 형식(0,2,3,4,5)으로 저장한다. /seq 는 마커와 QTL 데이터를 동일한 파일에 저장하며, /snp_code 에서 사용하는 코드 체계를 따른다. /gen 세대수1 세대수2 세대수3… 명령어를 통해 특정 세대에 대해서만 유전자형 데이터를 저장하도록 지정할 수 있다. 주의할 점은, /binary 옵션과 /snp_code 옵션은 동시에 사용할 수 없다.
해당 명령어를 사용하면 두개의 파일이 출력되는데, 하나는 마커 데이터 파일로 “개체군 이름”_mrk_“반복번호”.txt 형식으로 저장되며(Figure 10, Table 3), 다른 하나는 QTL 파일로 “개체군 이름”_qtl_“반복번호”.txt 형식으로 저장된다(Figure 11, Table 4). 이 두 파일은 개체 ID, 각 유전자좌에 해당하는 두개의 대립유전자 정보를 포함한다.
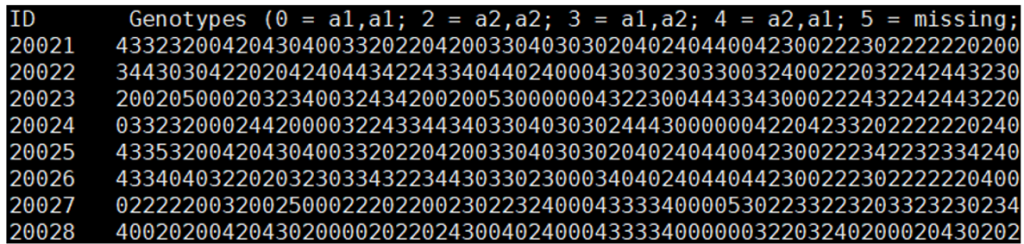
Figure 10. Example of a marker data file generated by the ‘genotype;’ command.
Table 3. Description of the marker data file generated by the ‘genotype;’ command.
| ID | Genotypes | 대립 유전자 정보 | |||
|---|---|---|---|---|---|
| 개체 ID | 유전자형 정보 | 0 : 동형접합(a1, a1) 2 : 동형접합(a2, a2) 3 : 이형접합(a1, a2) 4 : 이형접합(a2, a1) 5 : 유전자형 정보 없음 | |||
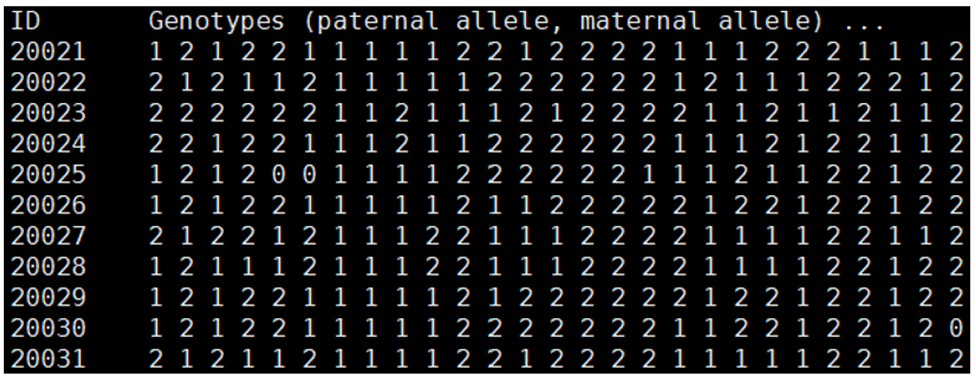
Figure 11. Example of a QTL data file generated by the ‘genotype;’ command
Table 4. Description of the QTL data file generated by the ‘genotype;’ command.
| ID | Genotypes (paternal, maternal) | 대립 유전자 정보 | |||
|---|---|---|---|---|---|
| 개체ID | 각 QTL에서 개체가 보유한 대립유전자 정보 (부계 및 모계 순서) | 11 : a1, a1 동형접합 | 22 : a2, a2 동형접합 | 12 혹은 21 : a1, a2 혹은 a2, a1 이형접합 | 0 : 데이터 없음 |
allele_freq option; 명령어는 대립유전자 빈도를 저장하는 기능을 하며, option 에는 /mafbin v 와 gen 세대수1 세대수2 세대수3 … 을 설정할 수 있다. /mafbin v 는 Minor Allele Frequency(MAF) 분포의 구간을 설정하는 역할을 하며 /gen 세대수1 세대수2 세대수3 … 은 특정 세대의 대립유전자 빈도를 저장하도록 하는 옵션이다. 결과 파일은 마커와 QTL 에 대해 두가지 형식으로 저장되며, 각각 “개체군이름_freq_mrk_반복수”.txt(Figure 12, Table5) 와 “개체군이름_freq_qtl_반복수”.txt(Figure 13, Table 6) 의 형태를 보인다.
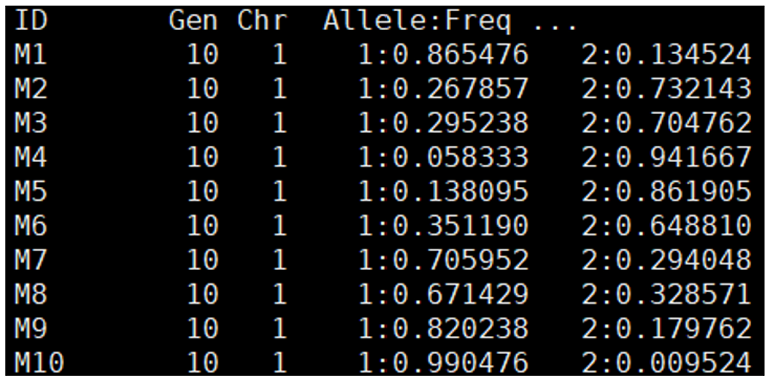
Figure 12. Example of a marker allele frequency file generated by the ‘allele_freq;’ command
Table 5. Description of the marker allele frequency file generated by the ‘allele_freq;’ command
| Marker ID | Gen | Chr | Allele 1 Frequency | Allele 2 Frequency |
|---|---|---|---|---|
| 마커 ID | 분석된 세대 | 염색체 번호 | 첫 번째 대립유전자가 나타날 확률 | 두 번째 대립유전자가 나타날 확률 |
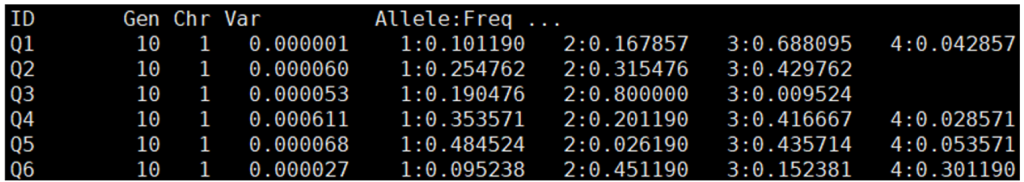
Figure 13. Example of a QTL allele frequency file generated by the ‘allele_freq;’ command.
Table 6. Description of the QTL allele frequency file generated by the ‘allele_freq;’ command
| QTL ID | Gen | Chr | Var | 대립유전자 빈도 |
|---|---|---|---|---|
| QTL ID | 분석된 세대 | 염색체 번호 | 변이값 해당 QTL에서의 변이 수준을 나타냄 |
하나의 QTL에는 최대 4개 이상의 대립유전자가 존재할 수 있다. |
Genome section은 개체군의 유전적 매개변수를 설정하여 유전체 정보를 정의하 구간이다. 해당 section은 크게 염색체를 정의하는 구간과(Figure 14), 추가적인 유전적 변이를 설정하는 구간으로 나누어진다(Figure 15).

Figure 14. Example of command structure in the Genome section for defining chromosome information and genetic parameters
begin_genome; & end_genome; 명령어를 사용하여 유전적 매개변수를 설정하는 구간의 시작과 끝을 정의한다. 이어서 begin_chr =value; & end_chr; 명령어를 사용하여 염색체의 정보를 정의한다. 여기서 value 는 염색체의 개수를 지정하며, 최대 500개의 염색체를 시뮬레이션 할 수 있다. 염색체마다 서로 다른 유전적 특성을 설정해야 할 경우, 각 염색체를 개별적으로 정의해야 한다.
chrlen = value; 명령어는 개별 염색체의 길이를 설정하는 항목이다. value 에는 1 ~ 10,000 범위의 값을 입력하며, 단위는 cM(Centimorgan) 이다. 이 때, 입력 값은 전체 염색체의 총 길이가 아니라, 각 염색체의 길이를 의미한다.
nmloci = value; 명령어는 개별 염색체 내의 마커 개수를 설정하는 항목이다. value 에는 0 ~ 400,000 범위의 값을 지정할 수 있으며, 시뮬레이션에서 마커 밀도를 결정하는 중요한 요소이다. 주의할 점은 전체 염색체의 마커 개수가 아닌, 각 염색체별 마커 개수를 의미한다는 것이다.
mpos option value; 는 마커의 위치를 설정하는 명령어이다. option 에는 even(균등하게 배치), rnd(무작위 배치), rnd1(첫 번째 반복에서만 무작위 샘플링 후, 동일한 마커 위치 유지)를 설정할 수 있으며, option 과 value에 mpos pd 위치1 위치2 위치3 …; 과 같이 설정하여 사용자가 마커 위치를 정의할 수 있다. 주의할 점은 정의한 마커 위치의 개수와 nmloci = value; 의 값이 반드시 동일해야 한다는 것이다. 예를 들어, mpos pd 1 5 23; 과 같이 세 곳에 마커를 배치한다면, nmloci = 3; 을 설정해야 오류가 발생하지 않는다.
nma = all num; 명령어는 첫 번째 historical 세대에서 마커 대립유전자 개수를 설정하는 항목이다. num 에 대립유전자 개수를 설정할 수 있으며, num = all 2; 로 설정할 경우 첫번째 historical population의 마커 대립유전자 개수는 2개로 설정이 된다.
maf = option; 역시 첫 번째 historical 세대에서의 마커 대립유전자 빈도를 설정하는 항목이다. option 값으로 eql(동일한 빈도), rnd(무작위로 샘플링된 빈도), rnd1(첫번째 에서만 무작위로 샘플링된 후 동일한 빈도)를 설정할 수 있으며, 이 설정은 첫 번째 historical 세대에서만 적용된다. 이후 세대에서는 유전적 부동(drift)과 돌연변이(mutation)에 의해 빈도가 변화할 수 있다.
nqloci = value; 명령어는 개별 염색체 내 QTL 의 개수를 설정하는 항목으로, value 값은 0 ~ 50,000 범위 내에서 지정할 수 있다. 이는 유전적 형질을 조절하는 주요 유전자 위치의 개수를 정의하는 요소로 작용한다.
qpos = option value; 는 QTL 위치를 설정하는 명령어이다. option 에는 even(균등하게 배치), rnd(무작위 배치), rnd1(첫 번째 반복에서만 무작위 샘플링 후, 동일한 QTL 위치 유지)를 설정할 수 있으며, option 과 value에 pd 위치1 위치2 위치3 … 과 같이 설정하여 사용자가 QTL의 위치를 정의할 수 있다. 주의해야 할 점은 정의한 QTL 위치의 개수와 nqloci = value;의 값이 반드시 동일해야 한다는 것이다.
nqa = all num; 명령어는 첫 번째 historical 세대에서 QTL 대립유전자 개수를 설정하는 항목으로, num에 대립유전자 개수를 설정할 수 있다. 이후 세대에서 돌연변이나 유전적 부동에 의해 대립유전자 개수가 변화할 수 있다.
qaf = option; 명령어는 첫 번째 historical 세대에서 QTL 대립유전자 빈도를 설정하는 항목이다. option 값에 eql(동일한 빈도), rnd(무작위로 샘플링된 빈도), rnd1(첫번째 에서만 무작위로 샘플링된 후 동일한 빈도)를 설정할 수 있으며, 이 설정은 첫 번째 historical 세대에서만 적용되고 이후 세대에서는 돌연변이와 유전적 부동의 영향을 받아 변화할 수 있다.
qae = option; 명령어는 첫 번째 historical 세대에서 QTL 대립유전자의 효과를 설정하는 항목이다. 사용자는 다양한 옵션을 통해 QTL 효과를 결정할 수 있다. qae = pd 분산값1 분산값2 분산값3 … 과 같은 옵션을 통해 QTL 분산 값을 개별적으로 정의할 수 있으며, 각 분산값의 합은 1이 되어야 한다. qae = rndg v; 를 설정하면 감마분포(gamma distribution)를 기반으로 효과를 샘플링 하며, v는 감마 분포의 모양 값(shape parameter)을 나타낸다. qae = rndn;을 설정하면 정규분포(normal distribution)에서 샘플링된 효과가 적용되며, qae = rnd; 옵션은 균등 분포(uniform distribution)에서 샘플링된 값이 QTL 효과로 적용된다. QTL 대립유전자 효과는 마지막 historical 세대에서 결정되며, 사용자가 설정한 분포에 따라 샘플링된 후 최종적으로 입력된 QTL 분산과 일치하도록 조정(scale)된다.
cld = option; 명령어는 첫 번째 historical 세대에서 완전한 연관불균형(Complete Linkage Disequilibrium)을 생성하는 옵션으로, 유전자 마커 및 QTL 간의 연관 불균형을 설정하는데 사용된다. option 에 따라 네 가지 방법으로 cld를 설정할 수 있는데, cld = m; 은 마커 간의 완전한 연관불균형을 생성하고, cld = q; 는 QTL간의 완전한 연관 불균형을 생성한다. cld = m q; 는 마커 및 QTL 간에 각각 독립적으로 연관불균형을 생성하지만, 마커와 QTL 간에는 LD가 적용되지 않는다. cld = mq; 는 마커간, QTL 간, 그리고 마커와 QTL 간 모두 완전한 연관불균형을 설정한다. 이 경우 마커와 QTL은 동일한 수의 대립유전자 수와 빈도를 가져야 하기 때문에, nma = all num;, nqa = all num;, maf = eql;, qaf = eql; 등의 설정이 필요하다. 또한, QTL 간의 완전한 LD를 설정하는 경우, recent population이 시작되기 전에 개체 간 변이를 생성할 수 있도록 historical 세대를 충분히 설정하는 것이 중요하다.
염색체 설정이 완료된 후, 마커 및 QTL의 위치를 무작위로 설정하거나, 돌연변이율과 결측률을 조정하는 등 추가적인 유전적 매개변수를 설정할 수 있다(Figure 15).

Figure 15. Example of additional parameter settings in the Genome section, including mutation rate and missing rate commands
r_mpos_g; 명령어는 각 반복 마다 마커 위치를 무작위로 설정하는 옵션이며, r1_mpos_g; 명령어는 첫 번째 반복에서만 무작위로 마커 위치를 설정한 후, 이후 반복에서는 동일한 위치를 유지하는 옵션이다. 마찬가지로 r_qpos_g; 명령어는 각 반복마다 QTL의 위치를 무작위로 배치하는 기능을 하며, r1_qpos_g; 명령어는 첫 번째 반복에서만 QTL 위치를 무작위로 설정한 후 이후 반복에서는 고정된 위치를 유지하는 옵션이다. 해당 옵션들에서 의미하는 반복은 GLOBAL PARAMETERS SECTION 의 nrep = num; 즉, 시뮬레이션 반복 횟수를 말한다.
rmmg = value /rndg v; 와 rmqg = value /rndf v; 명령어는 각각 마커 유전자형과 QTL 유전자형의 결측률을 설정하는 명령어이다. value 에는 0 ~ 0.5 사이의 결측률을 설정할 수 있으며, 추가적으로 /rndg v 옵션을 넣게 되면 감마분포를 이용해 결측률을 샘플링 할 수 있다. 여기서, v는 감마분포의 모양값을 나타낸다. 유전자형 결측은 시뮬레이션의 마지막 단계에서 적용되므로, 개체간 대립유전자 상속과정 에서는 영향을 미치지 않는다.
rmge = value /rndg v; 와 rqge = value /rndg v; 명령어는 각각 마커 유전자형과 QTL 유전자형의 오류율을 설정하는 항목이다. value 값은 0 ~ 0.2 범위에서 설정할 수 있으며, /rndg v 옵션을 추가하면 감마분포를 이용해 오류율을 샘플링 할 수 있다. 여기서 v 는 감마분포의 모양값을 나타낸다. 유전자형 오류는 개체군 내에서 임의로 대립유전자를 샘플링하여 발생하며, 원래의 올바른 유전자형과 다르게 설정된다. 샘플링된 대립유전자는 기존 개체군 내 존재하는 대립유전자에서 선택되며, 만약 특정 대립유전자가 고정된 상태라면 오류가 발생하지 않고 이형접합(heterozygous) 상태로 유지된다. 유전자형 오류는 시뮬레이션의 마지막 단계에서 적용되므로, 개체간 대립유전자 상속 과정에는 영향을 미치지 않는다.
mmutr = value option; 및 qmutr = value option; 명령어는 historical population 에서 마커와 QTL 돌연변이율을 설정하는 옵션으로, value 는 0 ~ 0.01 사이의 값을 가질 수 있다. 기본적으로 돌연변이 모델은 무한 대립유전자 모델을 따르며, 새로운 대립유전자가 생성되는 것이 아니라 기존의 대립유전자가 변경되는 방식으로 작동한다. option 에 /recurrent 를 사용할 경우 하나의 대립유전자만 존재하는 유전자좌는 이형접합으로 처리되어 시뮬레이션이 가능하도록 한다. 돌연변이 발생 횟수는 푸아송 분포(Poisson distribution)를 따르며, 개체 별 돌연변이 개수는 [u = 2 * 유전자좌 수 * 돌연변이 율]로 계산된 평균값을 이용하여 샘플링된다. 각각의 돌연변이는 유전체 내 무작위 유전자좌에 할당된다. recent population의 경우 세대수가 비교적 적어 돌연변이의 영향이 미미 하기 때문에 historical population의 돌연변이율을 설정하는 것이다.
Output Section은 시뮬레이션 결과를 저장하고 분석할 데이터를 선택하여 출력하는 구간이다(Figure 16).

Figure 16. Example of command structure for specifying output items to be saved from the simulation
begin_output; & end_output; 명령어를 사용하여 출력파일 매개변수를 설정하는 시작과 끝을 정의한다.
linkage_map; 명령어는 마커와 QTL 의 물리적 위치 정보를 포함한 파일을 출력한다. 해당 명령어를 사용하면 두개의 파일이 생성되는데, 마커의 연결지도는 “lm_mrk_반복 번호”.txt 형식으로 저장되며(Figure 17, Table 7), QTL의 연결지도는 “lm_qtl_반복 번호”.txt 형식으로 저장된다(Figure 18, Table 8).
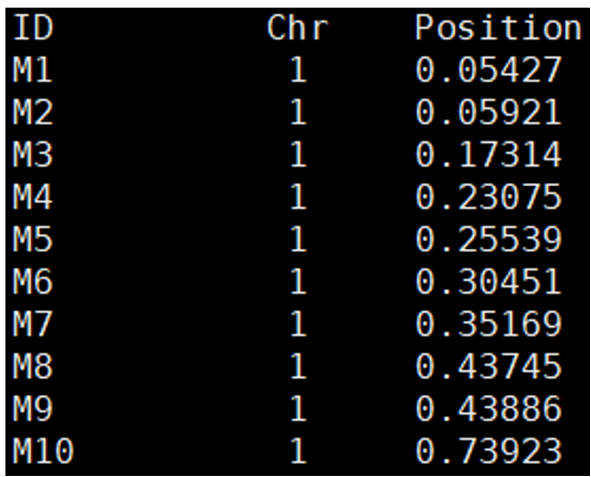
Figure 17. Example of a marker chromosome position file generated by the ‘linkage_map;’ command
Table 7. Description of the marker position file generated by the ‘linkage_map;’ command
| ID | Chr | Position |
|---|---|---|
| 마커의 ID | 해당 마커가 위치한 염색체 | 해당 염색체에서 마커의 위치 |
Figure 18. Example of a QTL position file generated by the ‘linkage_map;’ command
Table 8. Description of the QTL position file generated by the ‘linkage_map;’ command
| ID | Chr | Position |
|---|---|---|
| QTL ID | 해당 QTL이 위치한 염색체 | 해당 염색체에서 QTL의 위치 |
hp_stat; 명령어는 historical population의 간략한 통계정보를 저장하는 기능을 한다. “hp_stat_반복번호”.txt 형식의 파일이 생성되며 해당 파일 안에는 염색체 관련 정보, 유전자 밀도, 대립유전자 정보, 마커 및 QTL 돌연변이 수, 교차(crossover)정보, 세대별 개체군 정보가 포함되어 있다.
allele_effect; 명령어는 QTL의 효과크기 정보를 저장하는 기능을 가진다. “effect_qtl_반복번호”.txt 형식의 파일을 생성하며 염색체의 위치 및 대립유전자별 효과가 기록되어 있다(Figure 19, Table 9).
Figure 19. Example of a QTL allele effect size file generated by the ‘allele_effect;’ command
Table 9. Description of the QTL allele effect size file generated by the ‘allele_effect;’ command
| ID | Chr | Allele:Effect |
|---|---|---|
| QTL 식별 ID | QTL이 위치한 염색체 번호 | 각 대립유전자가 형질에 미치는 효과 |
monitor_hp_homo; 명령어는 historical population의 평균 동형접합도를 저장하는 기능을 한다. 옵션 /freq value를 사용하면 동형접합도를 특정 주기마다 기록할 수 있다. 예를 들어 value = 100 일 경우 100세대마다 데이터를 저장한다. 기본값은 50이며, 설정 가능한 범위는 1 ~ 10,000 이다. “hp_homo_mrk_반복 번호”.txt 파일은 마커의 평균 동형접합도를 저장하는 파일로, 첫번째 열(Gen)은 세대, 두번째 열(Mean homozygosity)은 해당 세대의 평균 동형접합도를 나타낸다(Figure 20). “hp_homo_qtl_반복 번호”.txt 파일은 QTL의 평균 동형접합도를 저장하는 파일로, 동일한 구조를 가지며 각 세대에서 QTL의 동형접합도를 나타낸다(Figure 21).
Figure 20. Example of a file showing average marker homozygosity by generation, generated by the ‘monitor_hp_homo;’ command.
Figure 21. Example of a file showing average QTL homozygosity by generation, generated by the ‘monitor_ hp_homo;’ command.
본 논문의 실습에서는 QMSim 프로그램의 설치 및 실행 방법을 다루고, 다양한 분석을 위한 매개변수 파일의 해석과 생성 과정을 상세히 설명하였다. 시뮬레이션 대상 축종은 Bos taurus로 설정하였으며, 29개의 염색체 (Womack, 1998), 형질의 유전력 (Lee and Kong, 2024), 그리고 cattleQTLdb (animalgenome.org/cgi-bin/QTLdb/index)에서 제공하는 데이터를 참고하여 매개변수 파일을 구성하였다. 이를 통해 QMSim 을 활용한 유전체 시뮬레이션을 보다 효과적으로 수행할 수 있도록 하는 것에 목표를 두었다.
프로그램 설치 및 실행은 Xshell Ver.8.0.0067 (NetSarang Computer Inc) 프로그램의 Linux 터미널 환경에서 진행하였다. QMSim은 Linux 환경에서만 이용할 수 있으며, 다음은 QMSim을 설치하고 실행하는 방법을 설명한다(Figure 22).

Figure 22. Instructions for installing and running the QMSim program
우선 QMSim 공식 홈페이지에 들어가 Download 항목 내 Linux version 의 링크주소를 복사한 후, Linux 터미널에 wget 명령어와 함께 붙여넣기 하여 다운로드 한다(Figure 23).

Figure 23. QMSim program successfully downloaded
이후 다운로드 된 QMSim 압축 파일을 unzip 명령어로 해제한다. 압축을 해제하게 되면 QMSim2_Linux 폴더가 생성되며, 해당 폴더에 들어가 QMSim 프로그램을 실행해본다. (Figure 24)와 같이 매개변수 파일의 입력하라는 문구가 나타나면, QMSim 프로그램의 설치는 끝난 것이다.
Figure 24. Message displayed upon running QMSim
매개변수 파일이 제작되었을 경우, (Figure 25)를 참조하여 QMSim을 실행한다.

Figure 25. Command to run the QMSim program with a parameter file
매개변수 파일에 이상이 없으면 오류없이 시뮬레이션이 진행되며(Figure 26), 생성된 폴더 내에 출력파일이 저장된다(Figure 27).
Figure 26. Successful execution of the simulation without errors
Figure 27. Output file
QMSim 매개변수 파일의 생성은 Window 환경의 메모장에서 진행되었다.
1. 선발 및 도태 시뮬레이션(Figure 28, Figure 29)

Figure 28. Selection and culling simulation – 1

Figure 29. Selection and culling simulation – 2
첫 번째 예제에서는 QMSim 프로그램의 기본적인 사용법을 이해하기 위해, 선발 및 도태 과정을 적용한 기초적인 시뮬레이션을 진행하였다.
Global parameter 에서 한우 도체중 형질의 총 유전력(h2)을 0.51로 설정하였으며 QTL 의 유전력은 총 유전력의 절반을 할당하였다.
Historical population section 에서 historical 세대의 개체군 크기는 3000마리에서 시작하여 1000세대 동안 1000마리로 축소된 이후 30세대 동안 530마리로 다시한번 축소되었다가, 20세대 동안 10000 마리로 확장하였다. 또한 마지막 historical 세대에서 수컷 개체 수를 5000마리로 고정시켰다.
Population section 에서는 historical 세대에서 수컷 20마리, 암컷 1,000마리를 무작위로 선발하여 founder를 구성하고, 20세대를 근친교배 최소화 교배방식으로 진행하였다. 선발은 추정 육종가가 높은 개체순으로, 도태방식은 표현형이 낮은 개체순으로 수행되었다. 매 세대 수컷은 50%, 암컷은 30%를 교체하였으며, BLUP방법을 이용하여 육종가를 추정하였다.
Genome section 에서는 염색체 29개를 설정하였으며, 각 염색체의 길이는 103 cM, 마커 수는 1,790개, QTL 개수는 118개로 지정하였다. 이에 따라 전체 유전체는 총 길이 2,987cM, 총 마커 수 51,910개, 총 QTL 수 3,422개로 구성된다. 또한, 마커 및 QTL에 대해 돌연변이율, 유전자형 누락률, 유전자형 오류율은 각각 2e-5, 0.01, 0.005로 설정하였다.
2. 서로 다른 방향의 두 계통 시뮬레이션 후 CrossBreeding (Figure 30, Figure 31)

Figure 30. Crossbreeding simulation example – 1

Figure 31. Crossbreeding simulation example – 2
두 번째 예제에서는 Population section에서 두 개 이상의 개체군을 설정하는 방법을 학습하기 위해, 서로 다른 기준으로 생성된 개체군 간의 교잡(Cross breeding) 시뮬레이션을 진행하였다. 이 외의 Section에서는 첫 번째 예제와 동일한 매개변수를 사용하였으므로, 이번 예제에서는 생략하였다.
Population Section에서 실제 육종가치가 높은 개체군 “POP1”과 실제 육종가치가 낮은 개체군 “POP2”를 생성한 이후, 수컷은 POP1, 암컷은 POP2의 각각 마지막 세대에서 추정 육종가가 높은 개체순서로 선발하여 교잡(Cross breeding)을 진행하였다.
3. 염색체 개별 시뮬레이션(Figure 32, Figure 33)

Figure 32. Chromosome-specific simulation example – 1

Figure 33. Chromosome-specific simulation example – 2
염색체마다 유전적 구조와 기능이 상이하기 때문에, QMSim 에서는 염색체의 유전적 매개변수를 개별적으로 정의할 수 있도록 설계되어 있다. 이를 통해 생물체의 염색체 간 차이를 정밀하게 반영할 수 있으며, 특정 염색체에 QTL이 집중되거나 거의 없는 영역을 설정함으로 유전적 Hotspot 과 Coldspot을 시뮬레이션 할 수 있다. 세 번째 예제는 염색체 별 유전적 매개변수를 직접 지정하여 시뮬레이션 하는 방법을 학습하기 위함으로, 이외의 Section은 첫 번째 예제와 동일한 매개변수를 사용하였기 때문에 생략하였다.
해당 예제에서는 3개의 염색체만 지정하였으며, 1번째 염색체의 경우 5780cM 의 길이, 2200개의 마커 수, 5개의 QTL 수를 할당하였다, 2번째 염색체는 3130cM의 길이, 8개의 마커, 6개의 QTL 수를 지정하였으며, 마커의 경우 mpos = pd … 명령어를 통해 마커의 위치를 설정하였다. 이어서 3번째 염색체는 4040cM 의 길이, 1600개의 마커, 7개의 QTL 수를 지정하였으며, qpos = pd … 명령어를 통해 7개 QTL의 위치를 설정하였다.
None.
This work was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (321082-3)
Philip K Thornton (2010). Livestock production: recent trends, future prospects, https://doi.org/10.1098/rstb.2010.0134
[DOI][PubMed][PMC]
FAO(Food and Agriculture Organization), 2020, The role of livestock in food security, poverty reduction and wealth creation in West Africa., FAO, Accra, West Africa
Goddard M, Hayes B, Meuwissen T. 2016. Genomic selection: A paradigm shift in animal breeding. Animal Frontiers 6(1):6-14.
[DOI]
Fernando Sc, Koyama M, Morota G, Silva Ff, Ventura Rv. 2018. Machine learning and data mining advance predictive big data analysis in precision animal agriculture. Journal of Animal Science 96(4):1540-1550.
[DOI][PMC]
Reents R, Stock Kf. 2013. Genomic selection: Status in different species and challenges for breeding. Reproduction in Domestic Animals 48(s1):2-10.
[DOI]
Vanraden Pm. 2020. Symposium review: How to implement genomic selection. Journal of Dairy Science 103(6):5291-5301.
[DOI]
Nayeri S, Sargolzaei M, Tulpan D. 2019. A review of traditional and machine learning methods applied to animal breeding. Animal Health Research Reviews 20(1):31-46.
[DOI][PubMed]
Chen Y, Gao H, Gao X, Guan L, Guo P, Li Jy, Niu H, Xu Ly, Zhang Jj, Zhang Lp, Zhu B. 2017. Effects of marker density and minor allele frequency on genomic prediction for growth traits in Chinese Simmental beef cattle. Journal of Integrative Agriculture 16(4):911-920.
[DOI]
Hall Sjg. 2016. Effective population sizes in cattle, sheep, horses, pigs and goats estimated from census and herdbook data. Animal 10(11):1778-1785.
[DOI][PubMed]
Astuti Pk, Bagi Z, Kichamu N, Kusza S, Strausz P, Wanjala G. 2023. A review on the potential effects of environmental and economic factors on sheep genetic diversity: Consequences of climate change. Saudi Journal of Biological Sciences 30(1):Article ID 103505.
[DOI][PubMed]
Amer Pr, Byrne Tj, Gibson Jp, Santos Bfs, Van Der Werf Jhj. 2017. Genetic and economic benefits of selection based on performance recording and genotyping in lower tiers of multi-tiered sheep breeding schemes. Genetics Selection Evolution 49:Article ID 10.
[DOI][PubMed][PMC]
Ahmadi A, Casellas J, Medrano Jf. 2010. Dairy cattle breeding simulation program: A simulation program to teach animal breeding principles and practices. Journal of Dairy Science 93(6):2816-2826.
[DOI]
Buttgen L, Ganesan A, Ha Nt, Pook T, Simianer H. 2021. A unifying concept of animal breeding programmes. Journal of Animal Breeding and Genetics 138(2):137-150.
[DOI][PubMed]
Dressel H, Koenig S, Scheper C, Swalve H, Wensch-dorendorf M, Yin T. 2016. Evaluation of breeding strategies for polledness in dairy cattle using a newly developed simulation framework for quantitative and Mendelian traits. Genetics Selection Evolution 48:Article ID 50.
[DOI][PubMed][PMC]
Lopes J, Mello Fcb, Oliveira Mmo, Rorato Prn. 2019. Strategies to control inbreeding in a pig breeding program: A simulation study. Ciência Rural. [in Portuguese]
[DOI]
Andonov S, Fragomeni Bo, Lourenco Dal, Masuda Y, Misztal I, Pocrnic I, Tsuruta S. 2017. Accuracy of breeding values in small genotyped populations using different sources of external information-A simulation study. Journal of Dairy Science 100(1):395-401.
[DOI][PubMed]
Battagin M, Edwards Sm, Faux Am, Gaynor Rc, Gonen S, Gorjanc G, Hearne Sj, Hickey Jm, Wilson Dl. 2016. AlphaSim: Software for breeding program simulation. The Plant Genome 9(3):Article ID plantgenome2016.02.0013.
[DOI][PubMed]
Reinhardt F, Simianer H, Täubert H. 2010. ZPLAN+: A new software to evaluate and optimize animal breeding programs.
https://www.unigoettingen.de/de/document/download/dc6158ed8a8d81851a97da84dbf9b561.pdf/t%C3%A4ubert.pdf
Bijma P, Rutten Mjmr, Van Arendonk Jam, Woolliams Jaw. 2002. SelAction: Software to predict selection response and rate of inbreeding in livestock breeding programs. Journal of Heredity 93(6):456-458.
[DOI][PubMed]
Sargolzaei M, Schenkel Fs. 2009. QMSim: A large-scale genome simulator for livestock. Bioinformatics 25(5):680-681.
[DOI][PubMed]
Aspilcueta-borquis Rr, Buzanskas Me, Guidolin Dgf, Matos Mc, Munari Dp, Nascimento Gb, Oliveira Hn, Seno Lo. 2018. Genomic selection in simulated dairy cattle populations. Journal of Dairy Research 85(3):272-277.
[DOI][PubMed]
Liu Y, Wang Y, Yin C, Yin Z, Zhou P. 2024. Using genomic selection to improve the accuracy of genomic prediction for multi-populations in pigs. Animal 18(2):Article ID 101062.
[DOI][PubMed]
Cardoso Ff, Costa Rf, Plotzki Reis A, Silva Ffe, Spangler Ml. 2019. The impact of selective phenotyping and genotyping over generations in beef cattle. Journal of Animal Science 97(Suppl_2):37-39.
[DOI]
Guo QiXin, Xu Lu, Lu Wei, Zhang KangNing, Yuan QingYan, Xu ShengHai, Wu Wei, Chang GuoBin, Chen GuoHong. 2016. Study on chicken breeding efficiency using GBLUP technique. China Poultry 38(20):6-9. [ref. 12]
Huang H, Li F, Wang Q, Xiang J, Yu Y, Zhang Q, Zhang X. 2019. Evaluation on the genomic selection in Litopenaeus vannamei for the resistance against Vibrio parahaemolyticus. Aquaculture 505:212-216.
[DOI]
Khalilisamani N, Khatkar Ms, Raadsma Hw, Thomson Pc. 2022. Estimating heritability using family-pooled phenotypic and genotypic data: A simulation study applied to aquaculture. Heredity 128:178-186.
[DOI][PubMed][PMC]
Womack Je. 1998. The cattle gene map. ILAR Journal 39(2-3):153-159.
[DOI][PubMed]
Kong Hs, Lee Gh. 2024. Estimation of genetic parameters and genetic evaluation of Hanwoo lean meat traits using BLUP. Journal of Animal Breeding and Genomics 8(4):207-215. [in Korean]
[DOI]