Hayeong Oh1†, Hyunji Choi2†, Yoonji Chung3, Phuong Thanh N. Dinh1, Youngjae Choi1, Jaeho Lee1, Woonyoung Jeong4, Seunghwan Ko1, JuHyeok Kim3, Seung Hwan Lee5*
1Department of Bio-AI Convergence, Chungnam National University, Daejeon, 34134, Korea
2Division of Animal Genomics and Bioinformatics, National Institute of Animal Science, Wanju, 55365, Korea
3Institute of Agricultural Science, Chungnam National University, Daejeon 34134, Korea
4Department of Bio-Big Data, Chungnam National University, Daejeon, 34134, Korea
5Division of Animal & Dairy Science, Chungnam National University, Daejeon, 34134, Korea
Correspondence to Seung Hwan Lee, E-mail: slee46@cnu.ac.kr
†These authors contributed equally to this work.
Volume 9, Number 1, Pages 29-37, March 2025.
Journal of Animal Breeding and Genomics 2025, 9(1), 29-37. https://doi.org/10.12972/jabng.2025.9.1.3
Received on January 17, 2025, Revised on March 04, 2025, Accepted on March 18, 2025, Published on March 31, 2025.
Copyright © 2025 Korean Society of Animal Breeding and Genetics.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0).

Classically, the way to find the information from papers is condensed as 1) search the papers through public search engines, 2) manually scan the textual data to obtain the information, such as gene, disease or protein. However, the number of published research is expanding at an increasing rate. Approximately 26 million papers were cited in PubMed in 2017, which is one of the most used databases of life science (https://www.nlm.nih.gov/bsd/licensee/baselinestats.html). Consequently, it makes a huge bottleneck to extract information by scanning all the paper manually. One of solution that can resolve this problem is Text-mining. In this study, we introduced computational step of Text-mining by R, a statistical language. Finding the genes associated with carcass weight was carried out as an example. In the results, the 786 papers were searched and the 236 genes were found in that papers, including NCAPG, MSTN, LCORL, POMC. The most frequently called gene related to cattle carcass weight is NCAPG(non-SMC condensin I complex subunit G).
Text-mining, Trait-association gene
형질 관련 유전자를 찾는 기존의 방법은 해당 형질과 관련된 논문을 검색하고 검색된 논문에서 직접 확인하는 일련의 과정을 거치기 때문에 연구자의 시간과 노력이 수반된다. 특히 생물·의학 데이터베이스인 PubMed에는 2017년 기준으로 약 2천 6백만 부의 논문이 수록이 되어 있고, 생물·의학 분야의 연구가 활발히 진행됨에 따라 그 수는 매년 증가하고 있다(https://www.nlm.nih.gov/bsd/licensee/ baselinestats.html). 증가하고 있는 많은 수의 논문 속에서 원하는 정보를 빠르게 추출하는 기술이 요구된다. 이때 사용할 수 있는 기술이 텍스트 마이닝이다 (Hearst 1997). 텍스트 마이닝 기법 중 개체명 인식 (named-entity reconition)은 문자형 데이터에서 찾고자 하는 카테고리(예를 들어 유전자, 단백질, 질병 등)의 단어를 추출하는 방법이다. 개체명 인식 방법에는 사전 기반 방식, 형태 기반 방식, 문맥 기반 방식 등이 있다 (Miguel 2011). 사전 기반 방식(Dictionary-Based)은 내가 찾고자 하는 단어들을 사전으로 제작한 후 해당 사전에 속하는 단어들을 텍스트에서 추출하는 방식을 말하며 높은 quality의 사전만 제작된다는 전제하에 정확한 단어의 추출이 가능하다. 이 방식은 사용자가 수동으로 사전을 제작해야 하는데, 현재 Ensembl database에는 인간, 소, 돼지 등 다양한 생명체의 유전자 정보를 제공하고 있어 사전 제작에 적합하다. 본 연구에서는 통계 프로그래밍 언어인 R(https://www.r-project.org/)을 활용한 사전 기반 개체명 인식 방법을 소개하고, PubMed 및 Ensembl 데이터베이스를 이용하여 소의 도체중과 관련된 유전자를 탐색하였다.
R 소프트웨어 4.4.0 버전을 이용 하였으며 이용한 라이브러리의 버전은 2024.12 기준 최신 버전을 이용하였다.
연구자가 원하는 정보만을 이용하기 위한 첫 단계는 R 환경에 관련 정보를 축적하는 것이다. NCBI-Entrez 데이터베이스 기반의 PubMed는 논문 및 문헌을 살펴볼 수 있는 서비스를 제공한다. PubMed를 형질 관련 유전자를 찾고자 하는 데 이용하고자 한다면 가장 먼저 분석하고자 하는 논문을 수집하게 되는데, 이때 PubMed 문헌 정보를 R에서 다룰 수 있도록 해주는 RISmed라는 패키지를 이용하여 홈페이지에 들어가지 않고 진행한다. 다음은 문헌 정보 검색을 위한 사전 준비 단계 코드이다.
install.packages(ʻRISmed’)
library(RISmed)
help(ʻRISmed’) #함수 기능 확인
query <- ‘carcass weight[TIAB]’ # 쿼리 생성
[TIAB] 는 PubMed 에서 제공하는 고급 검색 옵션으로 입력한 쿼리(ʻcarcass weight’)를 논문의 제목 혹은 초록에 포함하고 있는 논문만 검색하는 옵션이다. 자세한 옵션은 (https://www.ncbi.nlm.nih.gov/books/NBK3827/#pubmedhelp.Advanced_Search)에서 직접 확인이 가능하다. PubMed 의 데이터를 추출하고 생성하는 함수는 EutilsSummary 이며 다음 코드를 이용하여, query 정보로 논문을 끌어오고 str 함수를 이용해 추출한 article 객체를 확인하는 작업을 진행한다.
article <- EUtilsSummary(query, type=’esearch’, db=’pubmed’)
str(article)
Table 1. Formal class ‘EUtilsSummary’ [package “RISmed”] with 6 slots
| ..@ db | chr “pubmed” |
|---|---|
| ..@ count | num 2881 |
| ..@ retmax | num 1000 |
| ..@ retstart | num 0 |
| ..@ PMID | chr [1:1000] “39673410” “39647363” “39643874” “39639873” … |
| ..@ querytranslation | chr “\”carcass weight\”[Title/Abstract]” |
EutilsSummary 의 첫 번째 인자는 앞서 설정한 쿼리, 두번째 인자는 E-utilities의 종류이다. E-utilities 는 NCBI 데이터베이스 시스템을 다루기 위한 인터페이스이며 자세한 내용은 pubmed 홈페이지(https://www.ncbi.nlm.nih.gov/books/NBK25497/)에서 확인이 가능하다. 마지막 인자는 데이터베이스 종류이며 이번 예제에서는 PubMed 를 활용한다. str로 확인한 결과(Table1.)를 보면 총 6개의 슬롯으로 구성이 되어있는데, 쿼리에 대해 검색된 논문은 2,881편이지만 추출된 논문은 1,000편이라는 점에 주의한다. 만약 모든 논문을 전부 추출하고 싶다면 아래의 명령어를 이용하면 가능하다.
article <- EUtilsSummary(query, type=’esearch’, retmax=article@count, db=’pubmed’)
다만, retmax 의 기본값이 1000이며 사용자는 E-Utilities 서버의 오버로딩을 막기 위한 정책을 준수하여야 한다(https://www.ncbi. nlm.nih.gov/books/NBK25497/). 실제로 실행해보면, 호출된 모든 정보를 가져오는 것은 거의 불가능하다. 따라서 오류 없이 원하는 정보를 효율적으로 추출하기 위해서는 다음과 같이 query로 얻을 내용을 구체적으로 조정하는 것이 바람직하다.
query <- ‘carcass weight[TIAB] AND cattle[TIAB]’
AND로 연결하여 두 가지 조건을 만족하는 검색 결과를 가져올 수 있도록 조정하고, EUtilsSummary를 이용하여 pmid를 가져온다. str로 객체를 확인해 본 결과 오류 없이 정보를 가져올 수 있다고 판단할 수 있다.
article <- EUtilsSummary(query, type=’esearch’, db=’pubmed’)
str(article)
Table 2. Formal class ‘EUtilsSummary’ [package “RISmed”] with 6 slots
| ..@ db | chr “pubmed” |
|---|---|
| ..@ count | num 786 |
| ..@ retmax | num 786 |
| ..@ retstart | num 0 |
| ..@ PMID | chr [1:786] “39795001” “39790875” “39760134” “39736983” … |
| ..@ querytranslation | chr “\”carcass weight\”[Title/Abstract] AND \”cattle\”[Title/Abstract]” |
다음은 EutilsGet 함수를 이용하여 논문의 초록을 수집하는 방법이다. EutilsGet 함수는 EutilsSummary 로 검색된 PMID 를 이용해 실제 문헌 내용을 추출할 수 있게 해준다. AbstractText 함수로 생성된 abs 객체는 문자열로 이루어진 벡터이며 각 논문의 초록이 호출한 순서대로 벡터화되어 있다.
PMID <- article@PMID
article1 <- EUtilsGet(PMID,type=”efetch”, db=’pubmed’)
abs <- AbstractText(article1)
abs[1]
[1] ” … In this study, 20× whole-genome resequencing was performed on 282 Angus cattle from the Ningxia region, and a high-quality dataset encompassing extensive genomic variations across the entire genome was constructed. The iHS test identified 495 selection signal regions, which included pregnancy-associated glycoprotein ( PAG ) family genes and immune-related genes such as UL16-binding protein 21 ( ULBP21 ), CD1b molecule ( CD1B ), and tumor necrosis factor ligand superfamily member 11 ( TNFSF11 ). A quantitative trait locus (QTL) enrichment analysis revealed that several economic traits, including longissimus muscle area, marbling score, carcass weight, average daily gain, and milk yield, were significantly enriched in cattle with these selection signatures. ….”
벡터화되어 있는 데이터는 이용에 어려움이 있으므로 문장 간 줄 바꿈(\n) 기호를 추가하여 저장한다. 이때, 사용하는 cat 함수는 생성된 abs 객체를 .txt 형식의 파일로 저장하는 기능을 수행한다. 다음의 코드를 이용하여 가공한 초록 데이터를 저장한다.
abs[length(abs)] <- paste0(abs[length(abs)], ‘\n’)
cat(abs, file=’01.abstract.txt’)
저장된 초록의 예시는 다음과 같으며, 계속해서 논문들이 업로드되므로 예시로 제시된 결과와 다를 수 있다. 저장한 텍스트 형식의 파일은 R 환경의 현재 작업 디렉토리에서 확인이 가능하며, setwd 명령어로 디렉토리를 지정하고 getwd 명령어로 현재 작업 중인 디렉토리를 확인할 수 있다.

Figure 1. 저장된 초록 예시
지금까지 PubMed에서 원하는 형질과 관련된 유전자가 있다고 확인된 논문의 초록 텍스트를 수집하는 방법을 설명했다. 이 초록 텍스트 자료는 사용자가 필요로 하지 않는 정보까지 전부 합쳐져 있으므로 추가적인 가공이 필요하다. 따라서 초록에서 수집한 텍스트에서 필요한 정보만을 선별하기 위하여 Ensembl 데이터베이스를 이용한 유전자 사전 제작을 진행할 것이다. 해당 작업을 하기 위해 R의 ʻbiomaRt’ 패키지를 이용했으며, ʻbiomaRt’ 패키지는 Ensembl 데이터베이스로부터 데이터를 추출하는 인터페이스를 제공해준다. Bio-informatics 와 관련된 통계 tool을 관리하는 Bioconductor에서 제공하는 패키지로 다음의 코드로 설치 후 완료가 되면 패키지를 불러온다.
install.packages(‘BiocManager’)
BiocManager::install(‘biomaRt’)
library(biomaRt)
BiocManager를 이용했을 때 작업 환경에 따라 시간이 오래 걸릴 수 있으며, 모든 설치가 완료되었음을 확인하면, 유전자 리스트를 작성한다. ʻbiomaRt’패키지의 ’useMart’, ʻgetBM’ 함수를 이용하고, ʻUseMart’ 함수를 통해서는 사용하고자 하는 데이터베이스 및 데이터셋을 지정하는데, 앞서 NCBI-Entrez 데이터베이스에서 Bos taurus와 관련된 자료를 수집했으므로 “btaurus_gene_ensembl”를 사용했다. 데이터셋 지정을 완료했으면 ʻgetBM’함수를 사용해 유전자의 원하는 정보를 가져오면 된다. 가져온 결과에 대해 R의 ’tail’ 명령어를 이용하여 총 36,075개의 유전자 리스트가 작성된 것을 확인했다.
mart <- useMart(biomart=”ensembl”, dataset=”btaurus_gene_ensembl”)
whole_gene <- getBM(att ribute=c(“external_gene_name”,”ensembl_gene_id”,”description”), mart=mart)
tail(whole_gene)
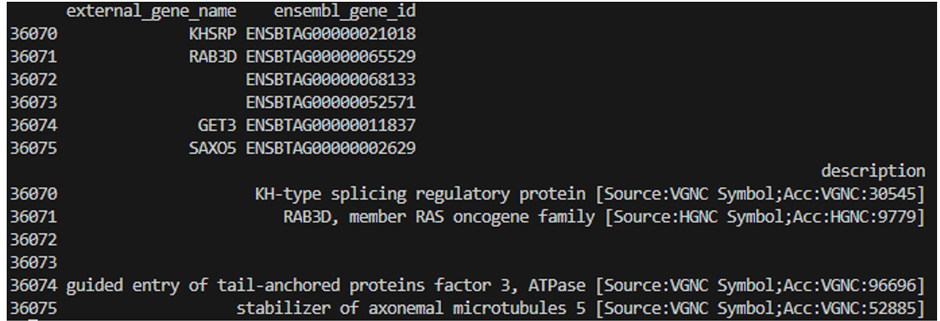
Figure 2. Ensembl database에서 추출된 36,075 유전자 리스트
사전기반 방식은 사전에 속해 있는 단어가 문맥에서 다른 의도로 사용이 되었을지라도 추출된다는 특징이 있다. 예를 들어 단어 ’ was’는 be 동사의 과거 단수형으로 문장에서 이용된다. 만약 유전자 사전에 Wiskott-Aldrich syndrome (WAS)이라는 gene이 존재한다면, 수집한 초록의 문맥에서 ’was’가 유전자로 추출된다. 의미에 맞지 않게 잘못 추출되는 오류를 범하지 않기 위해 텍스트 데이터에 추가적인 전처리가 필요하다. 다음 코드를 이용하여 문자열 데이터를 쉽게 다루기 위한 다양한 기능을 제공하는 ’stringr’를 설치 후 패키지를 불러온다.
install.packages(‘stringr’)
library(stringr)
인간유전자 명명법 위원회 (HUGO Gene Nomenclature Committee)의 Guidelines 에 따르면 현재 유전자 기호는 대문자 및 숫자로만 이루어져야 하므로 대문자와 숫자로 이루어진 단어만 추출하는 과정이 필요하다. 공백을 기준으로 단어 단위로 text를 나누고, 대문자 혹은 숫자를 포함하는 단어를 추출한 후 소문자와 특수문자를 지워주었다. 이런 코드를 이용하는 이유는 IGF2 와 같은 유전자를 추출하고자 할 때 IGF-2, IGF_2와 같은 방식으로 표기된 단어도 추출하기 위함이다. 조건에 맞는 유전자만 추출한 후, 추출된 단어의 빈도를 계산한다.
abs <- readLines(’01.abstract.txt’) #저장된 abstract.txt 파일을 불러옴
words <- strsplit(abs,’ ‘)[[1]] #공백을 기준으로 text 데이터를 단어 단위로 나눔
ity_words <- words[grep(‘[A-Z0-9]’,words)] #단어 내에 대문자, 숫자를 포함하는 단어를 추출
ity_words1 <- str_replace_all(ity_words,'[a-z]’,”) #추출된 단어에 속한 소문자 제거.
ity_words2 <- str_replace_all(ity_words1,'[[:punct:]]’,”) #추출된 단어에 속한 특수 문자 제거
#단어의 빈도 계산
count_ity_words <- table(ity_words2)
words_freq <- data.frame(word=names(count_ity_words), freq=c(unname(count_ity_words)))
words_freq$word <- as.character(words_freq$word)
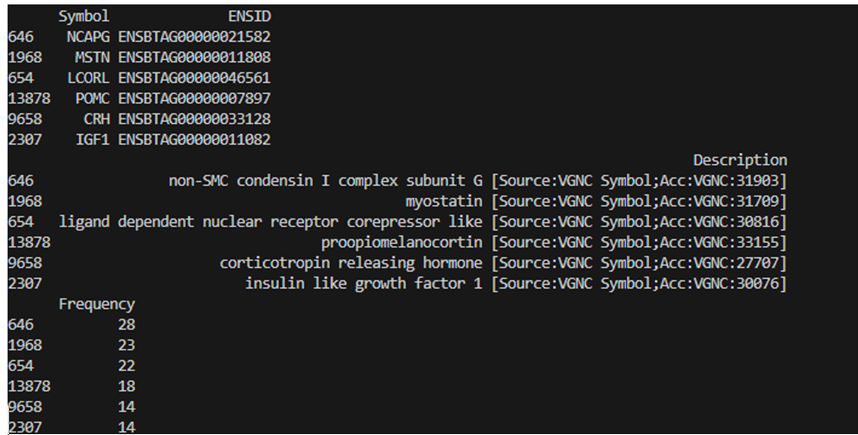
Figure 3. 정렬된 유전자 목록
텍스트 데이터에서 단어의 빈도까지 계산이 완료되면 다음 코드를 이용하여 전체 유전자 사전에서 Gene_symbol에 해당하는 내용만 추출한다. 추출된 단어를 매트릭스로 저장하기 위해 비어있는 myGeneList를 만들고, 이후 반복문을 통해 사전에 존재하고 글자 수가 3개 이상인 단어를 유전자로 인식해 빈도수와 사전정보를 가져와 비어있던 myGeneList에 붙여준다.
#앞서 생성한 유전자 사전에서 Gene_symbol 에 해당하는 영역만 가져온다.
gene_symbol <- whole_gene[,1]
#비어있는 메트릭스를 만들어 주고, 열이름을 지정해준다.
myGeneList <- matrix(NA,1,4)
colnames(myGeneList) <- c(‘Symbol’,’ENSID’,’Description’,’Frequency’)
for (i in 1:nrow(words_freq)) {
if (words_freq[i, 1] %in% gene_symbol & nchar(words_freq[i, 1]) >= 3) {
gene_information <- whole_gene[match(words_freq[i, 1], gene_symbol), ]
gene_information1 <- cbind(gene_information, words_freq[i, 2])
colnames(gene_information1) <- c(‘Symbol’, ‘ENSID’, ‘Description’, ‘Frequency’)
myGeneList <- rbind(myGeneList, gene_information1)
}
}
비어있는 첫 행을 삭제하고 유전자들을 빈도 순서대로 정렬해주는 코드로 결과를 수정한다. 추출된 유전자의 Symbol, 앙상블 ID, Description 및 발견빈도를 확인할 수 있고, 소의 도체중과 관련된 논문에서 가장 많은 빈도로 언급된 유전자는 NCAPG임을 확인했다. 또한, MSTN, LCORL, POMC 등의 유전자도 도체중과 밀접한 연관성을 보였다. 결과 파일은 텍스트 형식으로 저장하고, 저장이 완료된 파일은 사용자가 작업 중인 현재 작업 디렉토리에서 확인할 수 있다.
myGeneList <- myGeneList[-1,]
myGeneList <- myGeneList[order(myGeneList[,4], decreasing = T), ]
head(myGeneList)
write.table(myGeneList, ’02.GeneList.txt’, sep=’\t’, col.names = T, row.names = F, quote=F)
cat(paste0(‘Used query: ‘,query,’\n’,’number of articles: ‘,length(PMID),’\n’,’number of genes: ‘,nrow(myGeneList)),file=’summury.txt’)
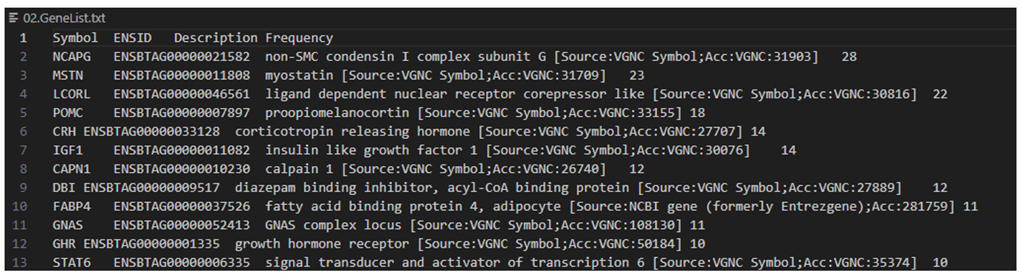
Figure 4. 탐색된 유전자 리스트
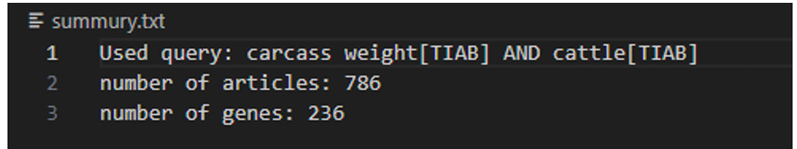
Figure 5. 결과 요약 파일 예시
마지막으로 R 패키지에서 텍스트 마이닝을 통해 얻은 결과를 효과적으로 시각화할 수 있는 워드 크라우드, bar plot 방법으로 시각화했다. 시각화에 사용하는 ʻwordcloud’ 그리고 ʻggplot2’ 패키지를 설치하고, 패키지를 불러온다.
install.packages(‘wordcloud’)
install.packages(‘ggplot2’)
library(wordcloud)
library(ggplot2)
패키지 설치를 오류 없이 완료하면 다음 코드를 이용해서 시각화를 진행한다. ʻpng’ 함수로 결과가 바로 그림 파일로 저장되도록 하고 ʻbrewer.pal’ 함수로 그림에 사용될 색을 지정한다. ʻwordcloud’ 함수는 첫 번째 변수에 단어의 벡터를 입력하고, freq 변수에 각 단어의 빈도를 넣어주고, scale 함수로 그림의 크기를 조절해주며 ʻmin.freq’ 변수로 그림에 나타낼 최소 빈도를 설정한다. 만약 ʻrandom. order’ 변수를 False로 지정하면 빈도가 높은 단어일수록 그림의 중앙에 위치하게 된다. 단어의 컬러는 ’random.color’를 True로 지정하여 다양하게 표현했다.
gene_freq <- myGeneList[,4]
names(gene_freq) <- myGeneList[,1]
png(file=”cloud.png”,height=900,width=1800,res=160)
palete <- brewer.pal(7,”Set3″)
palete[2] <- “#944EFF”
wordcloud(names(gene_freq), freq=gene_freq, scale=c(4,1), min.freq=5, colors=palete, random.order=F, random.color=T)
dev.off()
다음은 bar plot을 그리는 코드이다. 검출된 유전자의 빈도를 계산한 결과에서 상위 30개 유전자를 가져와 새로운 데이터프레임을 만든다. 워드크라우드와 마찬가지로 ʻpng’ 함수를 이용해 바로 그림 파일로 저장하며 ʻggplot’ 라이브러리를 이용해 plot을 그린다. ’ ggplot2’는 R에서 데이터 시각화와 관련된 강력한 내장기능을 포함하고 있으며 자세한 기능은 (https://ggplot2.tidyverse.org) 통해 확인할 수 있다.
rank_count <- data.frame(symbol=myGeneList[1:30,1],freq=myGeneList[1:30,4])
png(file=’chart.png’,height=900,width=1800,res=160)
ggplot(rank_count, aes(x = reorder(rank_count[,1], rank_count[,2]), y = rank_count[,2])) +geom_col() + coord_flip() + ylab(‘Frequency of genes’) + xlab(NULL) + scale_y_continuous(breaks = seq(0, rank_count[1,2], 5)) + theme_bw()
dev.off()

Figure 6. 워드 클라우드 시각화
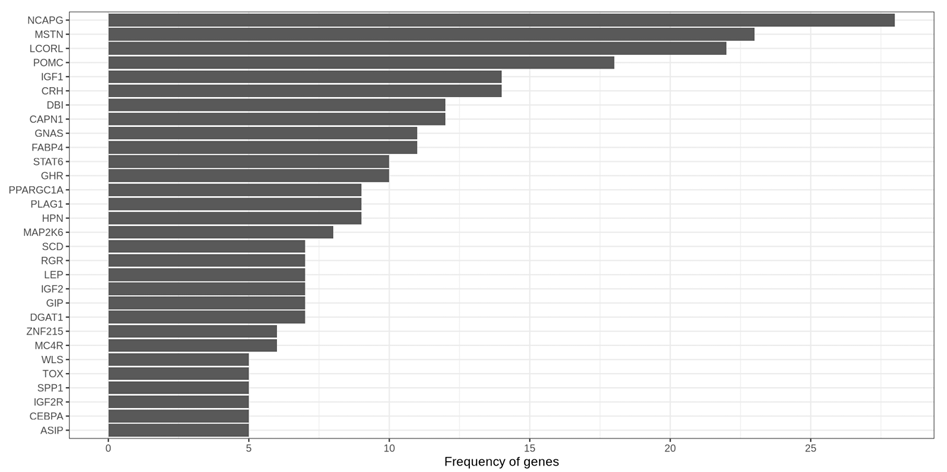
Figure 7. 탐색된 유전자 빈도 시각화
시간이 지남에 따라 데이터베이스의 크기가 방대해지면서 사람이 직접 정보를 찾는 데 한계가 있으므로 텍스트 마이닝을 이용해 연구자가 보다 효율적으로 정보를 얻을 수 있다. 본 연구는 텍스트 마이닝 기법을 활용하여 도체중과 연관된 유전자를 자동으로 추출하는 방법을 소개한다. NCBI-Entrez에서 도체중 관련 논문 748편을 추출하고, Ensembl 데이터베이스에서 36,075개의 유전자 리스트를 구축한 후, 사전 기반 개체명 인식 기법을 적용하여 236개의 형질 연관 유전자를 식별하였다. 사전을 이용해 확인한 결과 NCAPG유전자가 가장 높은 연관성을 보였다. 본 연구는 기존 수작업 방식에 비해 형질 연고나 유전자 탐색이 가능함을 보여주며, 향후 사람에 의해 수동적으로 추출된 유전자 명단이 있다면 텍스트 마이닝 기법의 성능평가 지표인 F1-Score 등을 활용하여 각기 다른 사전과 마이닝 방법에 따른 성능 비교가 가능할 것으로 기대된다.
None.
This work was supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (No. RS-2022-00155857, Artificial Intelligence Convergence Innovation Human Resources Development (Chungnam National University)).
This work was supported by the Technology Development Program (No.S3370836) funded by the Ministry of SMEs and Startups(MSS, Korea)
Miguel Vazquez, Martin Krallinger, Florian Leitner, Alfonso Valencia. 2011. Text Mining for Drugs and Chemical Compounds: Methods, Tools and Applications. Molecular Informatics, Volume 30, Issue 6‐7, 506-519. https://doi.org/10.1002/minf.201100005
[DOI][PubMed]
Hearst, M. A. 1997. Text data mining: Issues, techniques, and the relationship to information access. Proc. UW/MS workshop on data mining.
Stephanie Kovalchik. 2017. RISmed: Download Content from NCBI Databases. R package version 2.1.7. https://CRAN.R-project.org/package=RISmed
Steffen Durinck, Paul T Spellman, Ewan Birney, Wolfgang Huber. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols 4, 1184-1191.
https://doi.org/10.1038/nprot.2009.97
[DOI][PubMed][PMC]
Hadley Wickham. 2019. stringr: Simple, Consistent Wrappers for Common String Operations. R package version 1.4.0. https://CRAN.R-project.org/package=stringr
Ian Fellows. 2018. wordcloud: Word Clouds. R package version 2.6. https://CRAN.R-project.org/package=wordcloud
Hadley Wickham. 2016. ggplot2: Elegant Graphics for Data Analysis. Springer Cham.
https://doi.org/10.1007/978-3-319-24277-4_9
[DOI]
Sahadevan S, Hofmann-Apitius M, Schellander K, Tesfaye D, Fluck J, Friedrich CM, 2012. Text mining in livestock animal science: Introducing the potential of text mining to animal sciences. Journal of Animal Science, Volume 90, Issue 10, 3666-3676. https://doi.org/10.2527/jas.2011-4841
[DOI][PubMed]