Seunghwan Ko1†, Jeong Woen Shin1†, Yoonji Chung2, Phuong Thanh N. Dinh1, Young jae Choi1, Jaeho Lee1, Euiseo Hong3, Woonyoung Jeong3, Hayeong Oh1, JuHyeok Kim2, Seung Hwan Lee4
1Division Department of Bio-AI Convergence, Chungnam National University, Daejeon, 34134, Korea
2Institute of Agricultural Science, Chungnam National University, Daejeon 34134, Republic of Korea
3Department of Bio-Big Data, Chungnam National University, Daejeon, 34134, Republic of Korea
4Division of Animal & Dairy Science, Chungnam National University, Daejeon, 34134, Korea
Correspondence to Seung Hwan Lee, E-mail: slee16@cnu.ac.kr
†These authors contributed equally to this work.
Volume 9, Number 1, Pages 13-27, March 2025.
Journal of Animal Breeding and Genomics 2025, 9(1), 13-27. https://doi.org/10.12972/jabng.2025.9.1.2
Received on October 14, 2024, Revised on January 10, 2025, Accepted on February 24, 2025, Published on March 31, 2025.
Copyright © 2025 Korean Society of Animal Breeding and Genetics.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0).

Whole genome sequencing is a technology that detects nucleotide sequences across the entire DNA, which, in the past, was limited by high costs and time constraints. However, recent advancements in science and technology have significantly reduced these limitations. The GATK pipeline is widely used for detecting high-accuracy variants from whole genome data and offers the advantage of customizable variant filtering, making it a valuable tool in many research studies. This technical protocol provides a series of commands for practical use, from FASTQ data quality validation to variant detection and gene annotation using the GATK pipeline, employing the Bos taurus reference genome and whole genome sequencing data from five Hanwoo cattle obtained from the NCBI and SRA public databases. By presenting a method for high-quality variant detection, this paper is expected to contribute to improving the accuracy of downstream analyses, such as population studies and GWAS analyses.
Whole Genome Sequencing (WGS), Hanwoo SRA, GATK pipeline, Variant Calling, Annotation
DNA 시퀀싱 기술은 Sanger 시퀀싱으로 시작해 차세대 시퀀싱(NGS)으로 발전하며 전장 유전체 시퀀싱(WGS)을 가능케 했다 (Schuster 2008). 초기 NGS 시퀀싱은 긴 시간과 높은 비용의 한계를 가졌지만, 기술이 발전함에 따라 모든 측면에서 개선되었다(van Dijk, Auger et al. 2014). 현재 WGS는 24시간 이내에 $1,000 이하의 비용으로 수행 가능하며, 일부 플랫폼은 $500~$1,500까지 비용을 절감해 연구와 임상에서 실용성을 높이고 있다(Gullapalli, Desai et al. 2012).
현대 시퀀싱 기술을 주도하는 주요 기업으로는 Illumina, Element Biosciences, Thermo Fisher Scientific가 있으며 각기 다른 장단점이 존재하여 목적에 맞춰 사용되고 있다. Illumina는 NovaSeq 시리즈를 통해 대량 데이터를 효율적으로 처리하며(Kim, Jeon et al. 2021), Element Biosciences는 비용 효율적이고 간편한 소형 플랫폼을 제공한다. 마지막으로, Thermo Fisher Scientific의 Ion Torrent는 유전자 발현 분석과 표적 유전체 분석에서 강점을 보인다 (Michael A Quail, Miriam Smith et al. 2012).
이러한 WGS 데이터는 전장유전체에 걸친 단일염기다형성 (SNP)과 구조 변이 (SV)를 검출함으로써, SNP 칩의 한계를 넘어 높은 분석 정확도를 제공한다 (Song, Ye et al. 2019, Meuwissen, van den Berg et al. 2021). 뿐만 아니라, WGS 데이터에 대한 Genome-Wide Association Study (GWAS) 분석은 SNP array 데이터를 활용하였을 때 보다 더욱 많은 형질 연관 유전체 영역을 검출할 수 있었으며, 집단 분석에서도 높은 정확도를 보였다(Hoglund, Rafati et al. 2019).
WGS 데이터에 대한 변이 검출 도구로는 Samtools, Bcftools, VarScan, GATK 등이 있다(Koboldt, Chen et al. 2009, McKenna, Hanna et al. 2010, Danecek, Bonfield et al. 2021, Tung, Lien et al. 2021). Samtools와 Bcftools는 속도가 빠르고 메모리 효율성이 높아 간단한 변이 탐지에 적합하며(Danecek, Bonfield et al. 2021), VarScan은 낮은 빈도의 변이를 효과적으로 탐지한다(Koboldt, Chen et al. 2009). 마지막으로, GATK는 높은 정확도로 SNP, 구조적 변이(SV), 복제수 변이(CNV) 등 다양한 변이를 탐지한다(McKenna, Hanna et al. 2010). GATK의 표준화된 워크플로우는 데이터 정리, 품질 관리, 변이 호출 등 분석 전 과정을 체계적으로 지원함으로써 안정적이고 신뢰할 수 있는 결과를 제공한다(Tian, Yan et al. 2016, Bathke and Luhken 2021). 이 도구는 임상 유전학과 암 연구를 포함한 다양한 분야에서 활용되며, 1000 Genomes Project 같은 대규모 국제 연구에서도 표준으로 사용되고 있다.(McKenna, Hanna et al. 2010, Lee, Kweon et al. 2021).
본 논문에서는 GATK 파이프라인을 활용하여 WGS 데이터로부터 변이 추출, 품질 보정, 유전체 주석 정보 확보에 이르는 일련의 과정을 스크립트 및 옵션에 대한 자세한 설명과 함께 단계별로 제시하였다 (Figure 1). 또한, 한우 WGS 공개 데이터를 사용하여 데이터가 없어도 실습을 수행할 수 있도록 하였다.
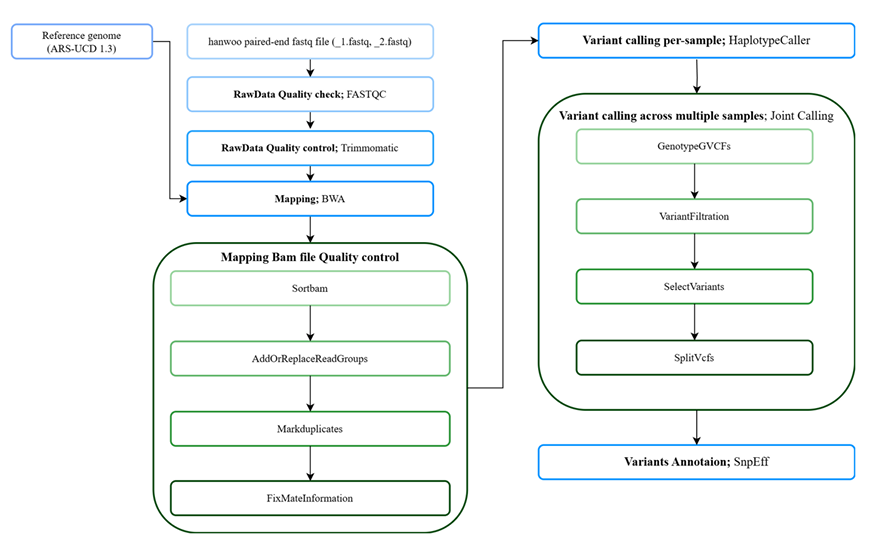
Figure 1. Graphical Abstract. This graphical abstract illustrates the steps of a variant detection pipeline. The process begins with rawdata quality control to assess and improve the quality of sequencing data. Mapping aligns reads to the reference genome, followed by BAM file quality control to ensure accurate alignment and data reliability. Next, variant calling identifies genetic variants for each sample, and joint calling combines data from multiple samples to improve variant detection accuracy. Joint calling file quality control refines the variants, and split variants separates them for detailed analysis. Finally, annotation provides functional information into the identified variants.
FASTQ는 시퀀싱하여 생상된 데이터 형식이다. 한 리드에 대하여 4줄로 작성되어 있으며, @로 시작하는 리드 이름, 염기서열, 구분자 (+), 그리고 ASCII code로 작성된 품질 점수 (Phread Score)로 구성되어 있다 (Figure 1). 이때, 시퀀싱 과정에서 다양한 오류와 편향이 생길 수 있기 때문에, 품질 관리 (Quality Control; QC)를 통해 고품질 데이터로 정제해야 한다. QC 과정에서는 가장 먼저 Raw Data의 품질을 시각화 하여 보정 여부를 결정하고, 어댑터 및 저품질 서열 필터링이 포함된다. 보통 Phred Score가 20보다 작은 리드는 분석 결과의 신뢰도를 저하시킬 수 있기 때문에 분석을 수행하기전 제거한다(Ewing and Green 1998).

Figure 2. FASTQ file format. The FASTQ file, which serves as the raw data format for whole-genome sequencing (WGS), consists of four lines per read: the read name, nucleotide sequence, separator, and quality score.
Mapping은 시퀀싱한 리드를 표준 유전체 (Reference genome)에 정렬하여 각 리드의 Reference genome 기반의 물리적 위치를 결정하는 단계이다(Li and Durbin 2009). Reference genome은 FASTA 형식으로 제공되며, 종에 대한 대표 유전체 서열이다. Mapping 과정에서는 Alignment algorithm을 사용하여 리드와 Reference genome 간의 일치 여부를 평가하고, 삽입 (Insertion) 및 결실 (Deletion)과 같은 작은 변이를 탐지할 수 있는 데이터를 생성한다(VanRaden, Bickhart et al. 2019). Mapping 결과 SAM 파일 형식으로 생성되지만, 압축 및 정렬 과정을 거쳐 BAM 파일 형식으로 변환하여 후속 분석에 사용된다.
Mapping 이후 생성된 BAM 파일은 변이 탐지의 기본 데이터로 사용되며, GATK는 이를 정제하고 품질을 보정하는 추가 QC 단계를 포함한다. 리드 그룹 할당, 중복된 리드 제거, INDEL 영역에 대한 재정렬 등과 같은 일련의 QC 과정은 변이 검출의 정확도 및 신뢰도를 확보한다(Adelson, Renton et al. 2019). 최적화된 BAM 파일은 변이 탐지 과정에서 오류 가능성을 줄일 뿐만 아니라 분석의 효율을 높여 전장 유전체에 걸친 고품질 변이를 확보할 수 있도록 한다.
GATK HaplotypeCaller는 보정된 고품질 BAM파일로부터 SNP 및 INDEL을 검출하는 도구이다. 이 과정은 총 다섯 단계로 구성되어 있다. 먼저, 변이가 발생할 가능성이 높은 영역을 Active Region으로 지정하여, 이 영역을 집중적으로 De Bruijn-like 그래프를 활용하여 잠재적인 Haplotype을 구성한다. 마지막으로, haplotype을 Reference genome과 비교하여 변이의 위치를 식별하고, Bayesian 접근법을 통해 유전자형 가능성 평가 후 변이 정보가 담긴 Genome Variant Call Format (gVCF) 형식으로 결과를 출력한다(Poplin, RuanoRubio et al. 2018).
Joint calling은 여러 샘플의 gVCF 파일을 동시에 처리하여 변이를 호출하여 하나의 VCF 파일로 통합시키는 단계이다. 이 과정에서 HaplotypeCaller 과정에서 생긴 noise를 제거하고, 일관적인 변이를 추출함으로써 고품질 변이 세트를 확보할 수 있다(Brouard, Schenkel et al. 2019). 이는 대규모 유전체 연구, 맞춤형 의료, 질병 유전자 연구 등에서 필수적인 단계로 활용된다(Chen, Boehnke et al. 2020).
변이 품질을 평가하고 불확실한 변이를 제거하는 과정으로, 분석 결과의 정확성을 높이는 것을 목적으로 둔다. GATK의 Variant Filtration 단계에서는 다양한 기준 (Mapping Quality, Quality Score, Depth of Coverage, etc.)을 적용하여 변이를 정량적으로 평가하고, 충족하지 못한 변이는 제거한다(Pirooznia, Kramer et al. 2014). 이를 통해 고품질 변이 데이터를 확보하고 최종적으로 분석 결과의 신뢰성을 극대화하는 것을 목표로 한다.
Annotation은 변이에 대한 주석 정보를 추가하여 생물학적 의미를 부여하는 단계로, 변이가 코딩 영역 또는 비코딩 영역에 위치하는지를 확인하고 그 기능적 영향을 평가한다(Cingolani 2012). 이 과정에서는 변이의 위치를 정확히 파악하고, 단백질 서열에 미치는 영향이나 유전자 발현 조절과 같은 기능적 변화를 예측한다. 이러한 주석 정보는 변이의 우선순위를 설정함으로써 유전체 데이터의 해석과 활용을 극대화하는데 필수적이다.
본 실습은 National Center for Biotechnology Information (NCBI) 데이터베이스로부터 Bos taurus Reference genome과 Sequence Read Archive (SRA) 데이터베이스로부터 한우 5개체에 대한 WGS 데이터를 다운받아 사용하였다 (Table 1). 실습은 총 14 단계로 구성되어 있으며, 각 단계에서 사용한 상세 옵션에 대한 설명은 Supplementary 1으로 제공되었다.
Table 1. Used data information
| Resource | Platform | Breed | Accession Number | Link |
|---|---|---|---|---|
| Reference genome | NA | Hereford | GCF_002263795.3 | https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/263/795/GCF_002263795.3_ARS-UCD2.0/ |
| Whole Genome Sequencing | Illumina HiSeq | Hanwoo | SRR934397 | https://www.ncbi.nlm.nih.gov/sra/?term=SRR934397 |
| Whole Genome Sequencing | SRR934433 | https://www.ncbi.nlm.nih.gov/sra/?term=SRR934433 | ||
| Whole Genome Sequencing | SRR934435 | https://www.ncbi.nlm.nih.gov/sra/SRX322372[accn] | ||
| Whole Genome Sequencing | SRR934436 | https://www.ncbi.nlm.nih.gov/sra/?term=SRR934436 | ||
| Whole Genome Sequencing | SRR934437 | https://www.ncbi.nlm.nih.gov/sra/?term=SRR934397 |
미니콘다 (Miniconda)는 생물정보학에서 자주 사용되는 경량화된 Python 배포판으로, 다양한 생물정보학 도구와 라이브러리를 설치하고 관리하기에 최적화된 환경을 제공한다. 주의해야할 점은 본인의 운영체제와 버전에 맞게 다운로드 해야 한다는 점이다. 다음 명령어는 Conda 환경을 구축하고, 본 실습에서 사용할 생물정보학 프로그램을 해당 환경에 설치하는 과정이다. 설치된 프로그램 정보는 Table 2에서 확인할 수 있다.
Table 2. Used software information
| Software | Version | Resource link |
|---|---|---|
| Burros-Wheeler Alignment Tools (BWA) | 0.17.18 | https://bio-bwa.sourceforge.net/ |
| FastQC | 0.12.1 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Samtools | 1.21 | https://www.htslib.org/ |
| htslib | 1.21 | https://www.htslib.org/ |
| Genomic analysis toolkit (GATK) | 4.6.1.0 | https://gatk.broadinstitute.org/hc/en-us |
| Trimmomatic | 0.39 | http://www.usadellab.org/cms/?page=trimmomatic |
[Command]
[Conda environment setting]
# Download lastest Miniconda3
mkdir miniconda3
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O
miniconda3/miniconda.sh
cd miniconda3
chmod +x miniconda.sh
bash miniconda.sh
# ENTER를 눌러 저작권을 확인하며 내려간 뒤 yes를 눌러 동의
# conda 설치경로 설정
# 설치한 conda를 환경변수에 입력
miniconda3/bin/conda init
source ~/.bashrc
# 커맨드에 (base)를 확인하여 conda가 정상적으로 작동했는지 확인
# 만약 (base)가 나오지 않는다면 아래 명령어를 통해 conda 실행
conda activate
# conda가 실행되지 않으면 터미널 껐다가 키기
conda config –add channels defaults
conda update -n base -c defaults
conda conda config –add channels conda-forge
conda config –add channels bioconda
conda create -n ngs
conda activate ngs
[Download Softwares in conda environment]
conda install -c bioconda
conda install -c bioconda multiqc
conda install -c bioconda samtools
conda install -c bioconda snpeff
conda install -c bioconda trimmomatic
wget <https://githu b.com/broadinstitute/gatk/releases/download/4.6.1.0/gatk-4.6.1.0.zip>
unzip gatk-4.6.1.0.zip
FastQC (version 0.12.1)은 FASTQ 안의 ASCII 코드로 암호화된 Phred score를 시각화하여 시퀀싱 데이터에 대한 품질을 한눈에 확인할 수 있게 한다. FastQC는 각 FASTQ 파일에 대해 독립적으로 수행된다.
[Command]
fastqc — threads ${Thread Number} ${FASTQ}
FastQC 결과, 각 리드의 영역에 따른 품질 통계량이 노란색 Boxplot으로 표시되며 빨간색 영역에서 초록색 영역에 위치할 수록 품질이 우수함을 나타낸다 (Figure 3).
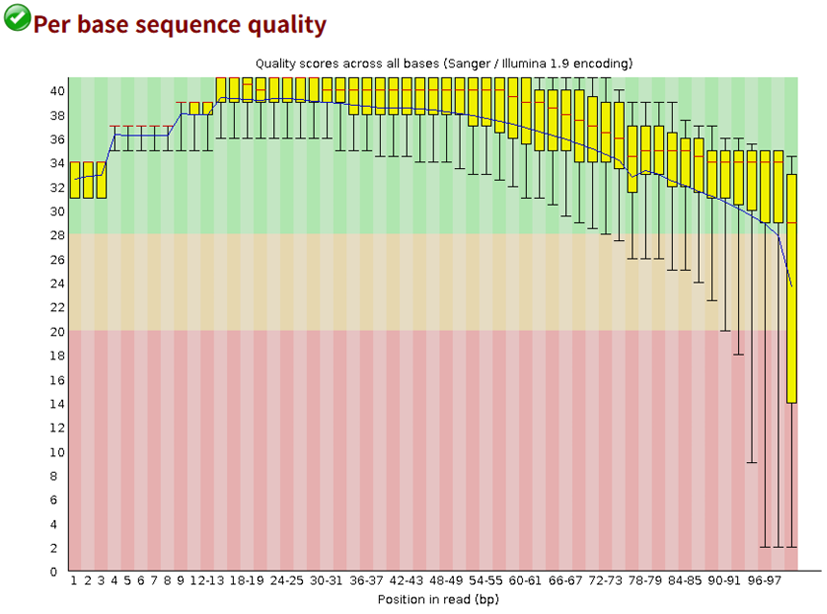
Figure 3. FastQC per base sequence quality result. The per-base quality statistics of reads in the FASTQ file are represented as yellow boxplots. The quality decreases as the data transitions from the green region to the red region, indicating lower read quality.
Trimmomatic (version 0.39)은 FASTQ 데이터의 품질을 보정하는 프로그램이다. Adapter 종류 및 품질 필터링 기준을 데이터 및 분석 목적에 맞게 설정할 수 있는 장점이 있으며, Paired-end 데이터인 경우 각 리드에 대한 Paired 및 Unpaired 고품질 리드 FASTQ 데이터를 출력한다.
[Command]
trimmomatic PE -threads ${Thread Number} -phred33 ${FASTQ_Read1} ${FASTQ_Read2} \
${PREFIX}_1.trimmed.P.fastq.gz \
${PREFIX}_1.trimmed.U.fastq.gz \
${PREFIX}_2.trimmed.P.fastq.gz \
${PREFIX}_2.trimmed.U.fastq.gz \
ILLUMINACLIP:${ADAPTER}:2:30:10:5:true \
LEADING:3 \
TRAILING:3 \
SLIDINGWINDOW:4:15 \
MINLEN:36
Burrows-Wheeler Alignment Tools (version 0.7.18)은 품질 보정이 완료된 리드에 대하여 Reference genome 기반 위치 정보를 확보하는 리드 맵핑 프로그램이다.
[Command]
bwa mem -M \
-t ${Thread Number} \
-R @RG\\tID:Bos_Taurus\\tSM:Bos_Taurus\\tPL:ILLUMINA \
${REFERENCE} \
${PREFIX}_1.trimmed.P.fastq.gz \
${PREFIX}_2.trimmed.P.fastq.gz \
| samtools sort \
-@ ${Thread Number} \
-o ${PREFIX}.bam
Mapping 결과 SAM 파일은 맵핑 결과에 대한 정보를 포함하고 있으며, 필수적으로 포함되는 11개 필드 (QNAME, FLAG, RNAME, POS, MAPQ, CIGAR, RNEXT, PNEXT, TLEN, SEQ,QUAL)와 선택적으로 추가될 수 있는 태그 필드로 구성되어 있다 (Figure 4). BAM 파일의 경우 이진화된 파일로 직접 확인할 수 없으며, ’samtools view’와 같은 프로그램으로 볼 수 있다.

Figure 4. SAM/BAM file format after BWA mapping. The SAM/BAM file, which contains the mapping information of reads, consists of 11 columns. From the first to the last column, the file includes: the unique read identifier, read status information, chromosome name in the reference sequence, starting position of the read, mapping quality score, string representing alignment information, name of the reference sequence for the read, position of alignment, length from end to end of paired reads, nucleotide sequence, and Phred quality score for the read.
GATK SortSam (version 4.6.1.0)은 Mapping 단계에서 생성된 BAM 파일을 염색체와 위치(coordinate) 기준으로 정렬하는 도구이다. 이를 통해, 탐색 및 작업 효율성과 변이 탐지 및 후속 분석의 정확도를 향상시킨다.
[Command]
/usr/bin/java \
-Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
SortSam \
–INPUT ${PREFIX}.bam \
–OUTPUT ${PREFIX}.sorted.bam \
–SORT_ORDER coordinate
GATK AddOrReplaceReadGroups (version 4.6.1.0)는 정렬된 BAM 파일에 리드 그룹 정보를 새로 추가하거나 기존 정보를 수정하는 단계다. 이 과정에서 리드 그룹 ID와 샘플 이름은 고유하게 설정해야 하며, 여러 파일을 병합할 때 충돌이 발생하지 않도록 주의해야 한다.
[Command]
/usr/bin/java \
-Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
AddOrReplaceReadGroups \
–INPUT ${PREFIX}.sorted.bam \
–OUTPUT ${PREFIX}.sorted.add.bam \
–RGLB ${PREFIX} \
–RGPL ILLUMINA \
–RGPU NONE \
–RGSM ${PREFIX} \
–SORT_ORDER coordinate
GATK MarkDuplicates는 리드 그룹 정보가 포함된 BAM 파일에 대해 중복된 리드를 식별하고 제거하는 도구다. 중복 리드는 PCR 과정에서 발생하거나 동일한 DNA 조각이 여러 번 시퀀싱될 때 생성된다. 이를 제거하면 변이 분석에서 발생할 수 있는 편향(bias)을 최소화하고, 결과의 신뢰도를 높일 수 있다.
[Command]
/usr/bin/java -Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
MarkDuplicates \
–INPUT ${PREFIX}.sorted.add.bam \
–METRICS_FILE ${PREFIX}.sorted.markduplicates.metrics.txt \
–OUTPUT ${PREFIX}.sorted.markduplicates.bam \
–ASSUME_SORT_ORDER coordinate \
–MAX_FILE_HANDLES_FOR_READ_ENDS_MAP 1024 \
–REMOVE_DUPLICATES true \
–DUPLICATE_SCORING_STRATEGY SUM_OF_BASE_QUALITIES \
–TAG_DUPLICATE_SET_MEMBERS true \
–REFERENCE_SEQUENCE ${REFERENCE}
GATK FixMateInformation은 중복 리드가 제거된 BAM 파일에서 쌍으로 생성된 리드의 Mate 정보를 정리하고 수정하는 단계다. BAM 파일 내의 페어링 관련 필드(예: mate reference name, mate position, insert size 등)가 잘못 기재되어 있으면, 분석 과정에서 오류가 발생할 수 있기 때문에 보정되어야 한다.
[Command]
/usr/bin/java \
-Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
FixMateInformation \
–INPUT ${PREFIX}.sorted.markduplicates.bam \
–OUTPUT ${PREFIX}.sorted.markduplicates.fixmate.bam \
–ADD_MATE_CIGAR true \
–ASSUME_SORTED true \
–CREATE_INDEX true \
–REFERENCE_SEQUENCE ${REFERENCE}
GATK HaplotypeCaller는 맵핑 정보가 보정된 BAM 파일로부터 변이(variants)를 검출하기 위한 도구다. 그 결과, 입력된 BAM 파일에 존재하는 변이 정보를 gVCF 형식으로 출력한다. HaplotypeCaller는 개별 BAM파일에 대하여 독립적으로 수행되기 때문에, 샘플마다 개별적으로 수행되어야 한다.
[Command]
/usr/bin/java -Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
HaplotypeCaller \
–input ${PREFIX}.sorted.markduplicates.fixmate.bam \
–output ${PREFIX}.sorted.markduplicates.fixmate.g.vcf \
–reference ${REFERENCE} \
–max-alternate-alleles 6 \
–native-pair-hmm-threads ${Thread Number} \
–emit-ref-confidence GVCF \
–standard-min-confidence-threshold-for-calling 20.0 \
bgzip -@ ${Thread Number} ${PREFIX}.sorted.markduplicates.fixmate.g.vcf
tabix -p vcf ${PREFIX}.sorted.markduplicates.fixmate.g.vcf.gz
생성된 gVCF는 헤더 (Header)와 데이터 라인 (Data Lines)으로 구성되며, 헤더는 “##”으로 시작하는 메타데이터와 #CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, FORMAT, SAMPLE 등의 열 이름을 포함한다. 데이터의 각 행은 변이 정보를 포함하고 있다 (Figure 5). 이 변이에 대한 정보는 유전자형 (GT), Depth of Coverage (DP), Genotype Quality (GQ), Minimum Depth (MIN_DP), Phredscaled Likelihoods (PL)가 있다.
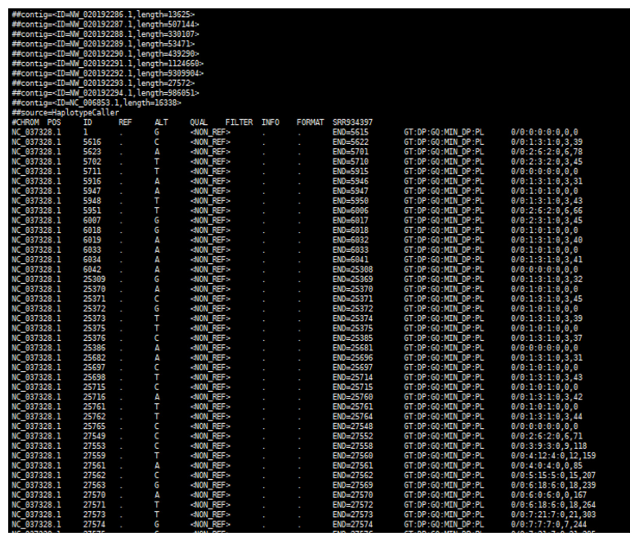
Figure 5. gVCF format after GATK HaplotypeCaller. The gVCF file, which contains variant information for an individual, consists of nine columns. From the first to the last column, the file includes: chromosome name in the reference sequence, variant position, unique identifier of the variant, reference base, alternative base, variant quality score, filtering status, additional information about the variant, and genotype information.
GATK GenotypeGVCFs는 여러 샘플의 gVCF 파일을 종합하여 최종 유전자형을 확보해주는 도구이다. Joint calling을 수행하고자 하는 모든 샘플에 대한 gVCF를 입력하면, 하나의 VCF 파일로 출력된다.
[Command]
/usr/bin/java -Xmx48g \
-jar gatk-package-4.6.1.0-local.jar \
GenotypeGVCFs \
–variant ${PREFIX1}.sorted.markduplicates.fixmate.g.vcf.gz \
–variant ${PREFIX2}.sorted.markduplicates.fixmate.g.vcf.gz \
–variant ${PREFIX3}.sorted.markduplicates.fixmate.g.vcf.gz \
–variant ${PREFIX4}.sorted.markduplicates.fixmate.g.vcf.gz \
–variant ${PREFIX5}.sorted.markduplicates.fixmate.g.vcf.gz \
–output Total.joint_calling.vcf \
–reference ${REFERENCE} \
–sequence-dictionary ${REFERENCE%.fna}.dict \
–max-alternate-alleles 6 \
–annotation-group StandardAnnotation \\
–annotate-with-num-discovered-alleles true
GATK VariantFiltration은 Joint calling 이후 하나로 통합된 VCF 파일을 다양한 필터링 기준에 따라 통과한 변이들에 대하여 Tagging 하는 과정이다. Input한 VCF와 변이 수 차이는 나지 않지만, 필터링 조건에 대한 만족 여부에 대한 정보가 추가된다.
[Command]
/usr/bin/java \
-Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
VariantFiltration \
–variant Total.joint_calling.vcf \
–output Total.joint_calling.filtered.vcf \
–reference ${REFERENCE} \
–sequence-dictionary ${REFERENCE%.fna}.dict \
–create-output-variant-index false \
–filter-name LowReadPosRankSum –filter-expression “ReadPosRankSum < -2.0” \
–filter-name LowMQRankSum –filter-expression “MQRankSum < -2.0” \
–filter-name LowQual –filter-expression “QUAL < 3.0” \
–filter-name QD –filter-expression “QD < 3.0” \
–filter-name FS –filter-expression “FS > 30.0” \
–filter-name MQ –filter-expression “MQ < 30.0” \
–filter-name DP –filter-expression “DP < 7” \
–genotype-filter-name DP –genotype-filter-expression “DP < 7” \
–genotype-filter-name GQ –genotype-filter-expression “GQ < 10.0”
bgzip -f -@ ${Thread Number} total.sorted.markduplicates.fixmate.filteration.vcf
tabix -f -p vcf total.sorted.markduplicates.fixmate.filteration.vcf.gz
GATK VariantFiltration에서 Tagging한 고품질 변이를 선발하는 과정이다. Output VCF파일에는 필터링 조건을 충족한 변이 세트로만 구성되어 있어, 후속 분석에 정확성과 신뢰성을 크게 향상시킨다.
[Command]
/usr/bin/java \
-Xmx40g \
-jar gatk-package-4.6.1.0-local.jar \
SelectVariants \
–variant Total.joint_calling.filtered.vcf \
–output Total.joint_calling.filtered.selected.vcf \
–reference ${REFERENCE} \
–sequence-dictionary ${REFERENCE%.fna}.dict \
–create-output-variant-index false \
–exclude-filtered true \
–exclude-non-variants true \
–set-filtered-gt-to-nocall true
bgzip -f -@ ${Thread Number} Total.joint_calling.filtered.selected.vcf
tabix -f -p vcfTotal.joint_calling.filtered.selected.vcf.gz
GATK SplitVcfs는 VCF 파일 내 변이 유형별(SNP, INDEL)로 데이터를 분리하는 과정이다. SNP와 INDEL로만 이루어진 2개 VCF 파일이 출력되며, 분석 목적에 따라 개별적으로 사용할 수 있다. 또한, 변이를 유형별로 구분함으로써 이후 분석이나 해석 과정을 보다 체계적으로 진행할 수 있다.
[Command]
/usr/bin/java \
-Xmx 128g \
-jar gatk-package-4.6.1.0-local.jar \
SplitVcfs \
–INPUT Total.joint_calling.filtered.selected.vcf.gz \
–INDEL_OUTPUT Total.joint_calling.filtered.selected.INDEL.vcf.gz \
–SNP_OUTPUT Total.joint_calling.filtered.selected.SNP.vcf.gz \
–STRICT false
snpEff는 변이에 대한 주석(Annotation)을 수행하는 도구로, 해당 변이가 유전자와 단백질 기능에 미치는 영향을 예측한다. 입력으로는 SplitVcfs 단계에서 생성된 VCF 파일이 주어지며, 출력으로는 주석 정보가 추가된 VCF 파일이 생성된다.
[Command]
# snpEFF에 Reference genome 데이터베이스 구축하기
vi snpEff.config
# 맨 아랫줄에 다음과 같이 줄을 추가하고 저장한다.
ARS_UCD2.0.genome : Bos_taurus_hereford
mkdir data ; cd data
mkdir ARS_UCD2.0 ; cd ARS_UCD2.0
cp GCF_002263795.3_ARS-UCD2.0_genomic.gff.gz genes.gff.gz
cp GCF_002263795.3_ARS-UCD2.0_genomic.fna.gz sequences.fna.gz
#snpEFF 실행
snpEff build -gff3 -noCheckCds -noCheckProtein -v ARS_UCD2.0
snpEff -c snpEff.config -v ARS_UCD2.0Total.joint_calling.filtered.selected.SNP.vcf.gz > snp.ann.vcf
snpEFF 결과, 입력한 VCF 파일에 각 변이에 대한 주석 정보 열이 추가된다. 주석 정보에는 변이가 속하는 유전자 이름, 유형, 효과, 영향 등급, 코딩 서열 위치, 단백질 변화, 영향받는 도메인 등의 정보가 포함되어 있다.
No potential conflict of interest relevant to this article is reported.
This work was supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government(MSIT) (No.RS-2022-00155857, Artificial Intelligence Convergence Innovation Human Resources Development (Chungnam National University)).
This work was supported by the Technology Development Program (No. S3370836) funded by the Ministry of SMEs and Startups (MSS, Korea)
Adelson, R. P., A. E. Renton, W. Li, N. Barzilai, G. Atzmon, A. M. Goate, P. Davies and Y. Freudenberg-Hua (2019). “Empirical design of a variant quality control pipeline for whole genome sequencing data using replicate discordance.” Sci Rep 9(1): 16156.
[DOI][PubMed][PMC]
Bathke, J. and G. Luhken (2021). “OVarFlow: a resource optimized GATK 4 based Open source Variant calling workFlow.” BMC Bioinformatics 22(1): 402.
[DOI][PubMed][PMC]
Brouard, J. S., F. Schenkel, A. Marete and N. Bissonnette (2019). “The GATK joint genotyping workflow is appropriate for calling variants in RNAseq experiments.” J Anim Sci Biotechnol 10: 44.
[DOI][PubMed][PMC]
Chen, Z., M. Boehnke and C. Fuchsberger (2020). “Combining sequence data from multiple studies: Impact of analysis strategies on rare variant calling and association results.” Genetic epidemiology 44(1): 41-51.
[DOI][PubMed][PMC]
Cingolani, P. (2012). Variant annotation and functional prediction: SnpEff. Variant calling: methods and protocols, Springer: 289-314.
[DOI][PubMed]
Danecek, P., J. K. Bonfield, J. Liddle, J. Marshall, V. Ohan, M. O. Pollard, A. Whitwham, T. Keane, S. A. McCarthy, R. M. Davies and H. Li (2021). “Twelve years of SAMtools and BCFtools.” Gigascience 10(2).
[DOI][PubMed][PMC]
Ewing, B. and P. Green (1998). “Base-calling of automated sequencer traces using phred. II. Error probabilities.” Genome research 8 3: 186-194.
[DOI][PubMed]
Gullapalli, R. R., K. V. Desai, L. Santana-Santos, J. A. Kant and M. J. Becich (2012). “Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics.” J Pathol Inform 3: 40.
[DOI][PubMed][PMC]
Hoglund, J., N. Rafati, M. Rask-Andersen, S. Enroth, T. Karlsson, W. E. Ek and A. Johansson (2019). “Improved power and precision with whole genome sequencing data in genome-wide association studies of inflammatory biomarkers.” Sci Rep 9(1): 16844.
[DOI][PubMed][PMC]
Kim, H. M., S. Jeon, O. Chung, J. H. Jun, H. S. Kim, A. Blazyte, H. Y. Lee, Y. Yu, Y. S. Cho, D. M. Bolser and J. Bhak (2021). “Comparative analysis of 7 short-read sequencing platforms using the Korean Reference Genome: MGI and Illumina sequencing benchmark for wholegenome sequencing.” Gigascience 10(3).
[DOI][PubMed][PMC]
Koboldt, D. C., K. Chen, T. Wylie, D. E. Larson, M. D. McLellan, E. R. Mardis, G. M. Weinstock, R. K. Wilson and L. Ding (2009). “VarScan: variant detection in massively parallel sequencing of individual and pooled samples.” Bioinformatics 25(17): 2283-2285.
[DOI][PubMed][PMC]
Lee, J. H., S. Kweon and Y. R. Park (2021). “Sharing genetic variants with the NGS pipeline is essential for effective genomic data sharing and reproducibility in health information exchange.” Sci Rep 11(1): 2268.
[DOI][PubMed][PMC]
Li, H. and R. Durbin (2009). “Fast and accurate short read alignment with Burrows-Wheeler transform.” Bioinformatics 25(14): 1754-1760.
[DOI][PubMed][PMC]
McKenna, A., M. Hanna, E. Banks, A. Sivachenko, K. Cibulskis, A. Kernytsky, K. Garimella, D. Altshuler, S. Gabriel, M. Daly and M. A. DePristo (2010). “The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.” Genome Res 20(9): 1297-1303.
[DOI][PubMed][PMC]
Meuwissen, T., I. van den Berg and M. Goddard (2021). “On the use of whole-genome sequence data for across-breed genomic prediction and finescale mapping of QTL.” Genet Sel Evol 53(1): 19.
[DOI][PubMed][PMC]
Michael A Quail, Miriam Smith, Paul Coupland, Thomas D Otto, Simon R Harris, Thomas R Connor, Anna Bertoni, Harold P Swerdlow and Y. Gu (2012). “A tale of three next generation sequencing platforms comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers.pdf.” BMC Genomics.
[DOI][PubMed][PMC]
Pirooznia, M., M. Kramer, J. Parla, F. S. Goes, J. B. Potash, W. R. McCombie and P. P. Zandi (2014). “Validation and assessment of variant calling pipelines for next-generation sequencing.” Human genomics 8: 1-10.
[DOI][PubMed][PMC]
Poplin, R., V. Ruano-Rubio, M. A. DePristo, T. J. Fennell, M. O. Carneiro, G. A. Van der Auwera, D. E. Kling, L. D. Gauthier, A. Levy-Moonshine, D. Roazen, K. Shakir, J. Thibault, S. Chandran, C. Whelan, M. Lek, S. Gabriel, M. J. Daly, B. Neale, D. G. MacArthur and E. Banks (2018). “Scaling accurate genetic variant discovery to tens of thousands of samples.” bioRxiv.
[DOI]
Schuster, S. C. (2008). “Next-generation sequencing transforms today’s biology.” Nat Methods 5(1): 16-18.
[DOI][PubMed]
Song, H., S. Ye, Y. Jiang, Z. Zhang, Q. Zhang and X. Ding (2019). “Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs.” Genet Sel Evol 51(1): 58.
[DOI][PubMed][PMC]
Tian, S., H. Yan, M. Kalmbach and S. L. Slager (2016). “Impact of post-alignment processing in variant discovery from whole exome data.” BMC Bioinformatics 17(1): 403.
[DOI][PubMed][PMC]
Tung, N. V., N. T. K. Lien and N. H. Hoang (2021). “A comparison of three variant calling pipelines using simulated data.” Academia Journal of Biology 43(2): 47-53.
[DOI]
van Dijk, E. L., H. Auger, Y. Jaszczyszyn and C. Thermes (2014). “Ten years of next-generation sequencing technology.” Trends Genet 30(9): 418426.
[DOI][PubMed]
VanRaden, P. M., D. M. Bickhart and J. R. O’Connell (2019). “Calling known variants and identifying new variants while rapidly aligning sequence data.” J Dairy Sci 102(4): 3216-3229.
[DOI][PubMed]