Department of Animal Science and Technology, Chung-Ang University, Anseong, Gyeonggi-do 17546, Republic of Korea
Correspondence to Jun-Mo Kim, E-mail: junmokim@cau.ac.kr
Volume 9, Number 1, Pages 1-12, March 2025.
Journal of Animal Breeding and Genomics 2025, 9(1), 1-12. https://doi.org/10.12972/jabng.2025.9.1.1
Received on October 14, 2024, Revised on January 10, 2025, Accepted on February 24, 2025, Published on March 31, 2025.
Copyright © 2025 Korean Society of Animal Breeding and Genetics.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0).

Pork consumption continues to increase worldwide, but with the acceleration of global heating, pigs are affected by heat stress that leads to reduce animal productivity. Although solutions of heat stress environments have been reported in various fields, research on transcriptome-based physiological changes is not enough. In this study, differentially expressed gene (DEG) profiling and gene ontology (GO) terms were identified through RNA-Seq analysis using whole blood of pig, categorized by the presence or absence of heat stress. As a result of DEG profiling, 354 genes were differentially expressed in week 1, and 99 genes were differentially expressed in week 2. In week 1, the TLR9 included in Toll-like receptor 9 signaling pathway was identified as a biological process (BP). Also, NOTCH1 was identified, which has an immunosuppressive effect by promoting and increasing TGF-β signaling. And in week 2, ALAS2 and FECH were identified as significant genes commonly involved in BP and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. As a result of functional enrichment analysis, candidate genes specifically expressed in pig heat stress environment were identified. This study provides insight into comprehensive understanding of heat-stressed pigs from a transcriptomic perspective.
Finishing pig, Heat stress, RNA-Seq, Differentially expressed gene, Functional enrichment analysis
한국의 축산업은 빠르게 성장하고 있으며, 육류 소비는 지속적으로 증가하고 있다. 전체 육류 소비량의 50% 정도를 차지하는 양돈산업은 축산업의 많은 부분을 차지한다 (KERI, 2023). 이에, 돼지 사육에 최적화된 환경을 맞춰주는 것이 동물 생산성의 증대를 위해 필수 요소이다. 하지만, 한국은 지구 온난화가 가속화되면서 기온의 증가 경향이 연중 뚜렷하게 나타나며, 덥고 습한 환경으로 변하는 중이다 (Jung et al., 2002). 주변 온도 및 습도의 증가에 따라 돼지가 고온 스트레스를 받는 것은 양돈산업에서 해결해야 할 문제 중 하나이다. 기후 변화로 인해 고온 스트레스는 특히 여름철에 동물의 건강과 성장에 영향을 미치는 주요 요인이 되었다 (Huo et al., 2019).
돼지가 고온 스트레스를 받을 때 발생하는 두 가지 주요 문제들이 있다. 첫째, 면역력 저하로 인해 질병에 취약해진다 (Huo et al., 2019). 돼지는 땀샘이 잘 발달되어 있지 않고, 두꺼운 피하 지방 조직을 가지고 있다 (Wolp et al., 2012). 이 특성들을 가진 돼지들이 고온 스트레스를 받으면 장기에 각기 다른 영향을 미치며, 복사열 발산을 최대화하기 위해 대부분의 혈액을 피부로 분산하여 내부 고온을 체외로 발산한다 (Pearce et al., 2013). 혈액이 각 조직과 세포에 영양분과 산소를 공급해야 하는데, 고온 스트레스로 인해 제 역할을 하지 못해 내장 조직으로의 혈류와 산소 및 영양분 공급이 감소하게 된다 (Cantet et al., 2021). 이로 인해, 세포가 조직 내에서 균열을 일으켜 각종 세균과 바이러스가 혈류에 침투해 돼지에게 염증이나 질병을 일으킨다. 둘째, 돼지의 사료효율과 성장률이 감소하게 된다. 돼지는 경제동물 중 큰 비중을 차지하며, 양돈산업에서 경제적 가치를 도출하려면 기본적으로 사료효율과 성장률을 높여야 한다. 그러나 돼지가 고온 스트레스를 받으면 생리적 조절이 어려워지기 때문에 섭취하는 사료에서 발생하는 대사열을 줄이기 위해 자발적으로 사료 섭취량을 줄인다 (Witte et al., 2000). 또한, 돼지는 평소보다 더 많은 에너지를 소모하게 되어 사료 효율이 떨어지게 된다 (D. L. Ingram, 1965). 이처럼 고온 스트레스는 양돈 산업의 경제적 측면에서 부정적 영향을 미쳐 해결책 모색이 시급하다.
하지만, 이에 대한 대책은 돈사 내 냉각 시스템 및 선풍기 설치 등 외부적인 해결 방법만 있을 뿐이다 (Godyń et al., 2020). 더불어, 고온 스트레스 환경에서 발생하는 돼지의 생리학적 변화를 파악하고자 하는 노력이 다방면으로 시도되고 있으나, 전사체 관점에서의 생리학적 변화에 대한 연구는 타 축종에 비해 부족한 것이 실정이다 (Luo et al., 2021; Perini et al., 2021). 차세대 염기서열분석 (NGS, Next Generation Sequencing) 기술이 발달함에 따라 많은 데이터를 저렴하고, 신속하게 생산할 수 있어 전사체에 대한 연구를 빠르게 변화시켰다 (Hrdlickova et al., 2017; Patel and Jain, 2012). 유전자 발현의 차이를 볼 수 있는 전사체 데이터로 유전자 발현 분석을 하여 생명현상에 대한 통찰력을 얻을 수 있다 (Harrison et al., 2012; Melé et al., 2015). 또한, 생물체는 처한 환경마다 각기 다른 유전자 발현 네트워크를 이루며 살아가기 때문에 유전자 발현의 연구를 통해 생물학적 과정을 심층적으로 이해할 수 있다 (Marguerat and Bähler, 2010). 따라서, 본 연구는 만성적 고온 스트레스의 효과 확인하기 위하여 2주간 고온 스트레스를 부여함으로써 각 주령별 조건 하에 조직과 세포에 산소 및 영양분을 공급 역할을 하는 전혈에서 전장 전사체를 추출하였다. High-throughput 특성을 지닌 NGS 기술로 분석한 전사체 데이터를 이용하여 시점 별 차등발현유전자 (DEG, Differentially Expressed Gene)들을 식별하였고, 생물학적 기능 분석을 통하여 고온 스트레스로 인한 비육돈의 반응 기작에 대한 기전 구명 및 통찰력을 얻기 위해 본 연구를 수행하였다.
10마리의 비육돈[(Landrace x Yorkshire) x Duroc; initial body weight = 51.29 ± 9.35 kg]들이 실험에 사용되었다. 모든 실험과정은 동물실험윤리위원회(IACUC)의 승인을 받고 수행하였다(approval # 202012A- CNU-204). 5마리씩 그룹화하여 한 그룹은 대조군, 나머지 그룹은 실험군으로 구분 지었다. 선행연구에 근거하여 대조군은 온도를 23 ~ 24°C, 습도를 35%로 설정하였고, 실험군은 온도를 32 ~ 33°C, 습도를 80%로 처리하여 비육돈에 고온 스트레스를 부여하였다 (Xin and Harmon, 1998). 실험은 바닥이 100% plastic slatted, pen의 크기가 2.32m × 1.75m × 0.7m인 돈사에서 14일 동안 진행되었으며, 전체 실험기간 동안 사료 및 음수는 자유 급여하였다. 실험기간 중 1, 2주 차 때 각각의 돼지 샘플로부터 전혈을 채취하였다.
각 대조군과 실험군의 0주차의 초기 체중(BW, Body Weight), 2주차의 최종 체중, 평균 일당 증체량(ADG, Average Daily Gain), 평균 일당 사료 섭취량(ADFI, Average Daily Feed Intake) 및 사료 전환 효율(G:F, Gain:Feed Ratio) 총 5가지 경제적 형질에 대한 통계 분석을 SAS 9.4 통계 소프트웨어 패키지(SAS Institute Inc., Cary, North Carolina, USA)를 이용하여 수행하였다. 각 그룹 간의 차이는 독립 표본 T-test를 이용하여 분석하였으며, 그룹 간 평균 차이는 P-value 0.05 및 0.01 이하일 때 통계적으로 유의하다고 판단하였다.
TRIzol 시약(Invitrogen, Life Technology, Carlsbad, CA, USA)을 사용해서 제조업체의 권장사항에 따라 Total RNA를 전혈에서 분리하였다. NanoDrop ND-1000 분광광도계(NanoDrop Technologies, Wilmington, DE, USA)를 이용하여 RNA를 정량을 측정하고, 샘플의 상태를 확인하였다. Illumina TruSeq™ RNA Sample Preparation Kit로 Total RNA 중 mRNA 1 µg을 library로 제작하였다. mRNA를 분절화 과정으로 작은 조각으로 만들었고, 이를 역전사효소와 primer을 이용하여 cDNA의 첫 번째 가닥으로 만들었다. DNA polymerase 1과 RNase H를 이용하여 cDNA의 두 번째 가닥을 만든 후, end-repair과정을 진행하였다. Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA) 을 이용하여 paired-end (2 X 100 base pair) sequencing을 진행하였다.
각 대조군과 실험군에 대한 DEG들의 기능을 예측하기 위해 기능분석을 수행하였다. GO terms (Consortium, 2004)와 KEGG pathway (Kanehisa and Goto, 2000)의 해석을 하기 위해서 DAVID (Database for annotation, Visualization and Integrated Discovery) v2021 (Sherman et al., 2022)을 기능 해석 도구로 사용하였다. DAVID 기본옵션인 Count 2개 이상, EASE score (P-value) 0.1 이하를 cut-off 기준 값으로 이용하였다. GO 기능 분석은 biological process로 수행하였다. 그 후, REVIGO tool (Supek et al., 2011)을 이용하여 enriched GO term의 Treemap을 시각화하였다. KEGG annotation은 –log10P-value, fold enrichment로 표현하였다. 유전자 공동 발현 네트워크는 선별된 DEG들을 Cytoscape v3.10.0 (Shannon et al., 2003)의 plug-in인 ClueGo (Bindea Gabriela et al., 2009)와 CluePedia (Bindea G. et al., 2013)에서 P-value 0.05 이하를 cut-off 기준 값으로 이용하였고, Cluego parameter는 GO Term; min level = 3, max level = 8, genes; Min = 3, Min gene percentage = 4.000%으로 설정하여 결과값을 시각화하였다.
고온스트레스가 돼지의 생산성에 미치는 영향을 평가하기 위해 총 5가지의 경제적 형질을 측정하였다. 체중 변화, 일당 증체량 및 사료섭취량은 대조군에서 높았고, 사료 전환 효율은 실험군에서 높은 경향을 보였다 (Table 1).
Table 1. Effects of heat stress on productivity performance in finishing pig
| Group | Body Weight, kg | Average Daily Gain, kg/d** | Average Daily Feed Intake kg/d** | Gain:Feed Ratio (G:F) | |
|---|---|---|---|---|---|
| Week 0 | Week 2 | ||||
| Control | 54.31 | 65.41 | 0.80 | 1.90 | 0.42 |
| Treat | 51.78 | 58.67 | 0.49 | 1.15 | 0.43 |
*p < 0.05; **p < 0.01
실험군과 대조군 사이의 상관관계를 알아보기 위해서 10마리의 돼지들을 선택하였다. 선정된 각 돼지들을 대상으로 평소 환경을 적용한 대조군과 고온 스트레스를 가한 실험군을 비교 분석하였다. 먼저, RNA-Seq 데이터 전처리과정을 수행하여 샘플 별 trimming 비율, 매핑 비율 등을 나타내었다. Sample name에서 F는 1, 2주 차별 돼지를 구별한 것이고, T에서 T1은 일반 그룹, T4는 고온 스트레스를 받은 그룹으로 나눈 것이다. RNA-Seq 데이터 전처리 결과로 대조군과 실험군 각각에서 평균 39,813,498개의 raw read가 있었고, GC (Guanine-Cytosine)의 평균함량은 44.2%인 것으로 도출되었다. Trimming 과정 이후, 평균 39,060,464개의 read들이 있었고, GC의 평균함량은 44.2%였다. 매핑 이후, 고유한 위치에 특이적으로 매핑된 read들의 평균비율이 87.42%, 모든 위치에서 매핑된 read들의 평균비율은 96.7%이었다 (Table 2). 벤 다이어그램으로 주차 별 DEG들 간의 관계를 볼 수 있었다 (Fig 1A). MDS plot 및 Volcano plot은 주차 별 대조군 및 실험군 간 전사체 기반의 유전자 발현패턴을 나타내었으며, 샘플이 뚜렷하게 군집화된 것을 보여준다 (Fig 1B, C). 1, 2주 차 MDS plot에서 NC는 대조군, HS는 실험군을 의미하며, 대조군의 F1.T1.67.B, F2.T1.110.B, 실험군의 F2.T4.19.A, F2.T4.105.B는 x, y축에서 이상치로 나타나 이 샘플들을 제거하였다. MDS plot에서 대조군과 실험군의 분포를 확인하였다. Volcano plot에서는 DEG들의 임계치를 확인하고, 14일 동안의 DEG 발현패턴을 볼 수 있었다. 1주 차의 Volcano plot에서는 68개의 하향 조절 및 286개의 상향 조절된 DEG들을 발견했고, 2주 차의 Volcano plot에서 81개의 하향 조절 및 18개의 상향 조절된 DEG들을 발견했다. Volcano plot에서 주 차별 상향 및 하향 조절된 DEG들을 발견했고, 이 유전자들을 비교할 수 있었다. 2주 차는 1주 차와 달리 DEG들의 하향 조절된 수가 상향 조절된 수보다 많았으며, 1주 차에서 DEG들이 가장 많이 발현된 것을 확인할 수 있었다 (Fig 1B, C).
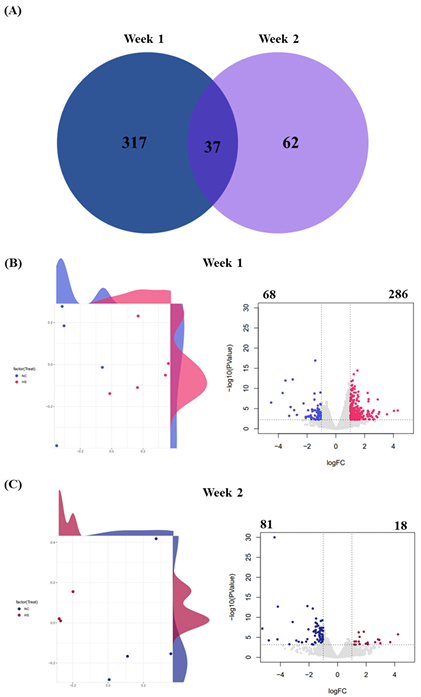
Figure 1. RNA-Seq transcriptional profiling in 2 weeks of each weeks to pigs. Venn diagram of DEGs in week 1 and 2 (A). MDS and Volcano plot of week 1 (B) and week 2 (C).
Table 2. Summary of RNA-Seq raw reads, trimming, and mapping to pig genome
| Group | Sample name | Raw | After trimming | After mapping | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Sequence length | GC(%) | Remaining reads after trimming | Sequence length trimmed(bp) | Trimmed GC(%) | Trimming rate(%) | Uniquely mapped rate(%) | Overall mapped rate(%) | ||
| Control | F1.T1.10.4.B | 43,914,182 | 101 | 44 | 43,109,613 | 36–101 | 44 | 1.83 | 83.74 | 96.77 |
| F1.T1.11.0.B | 44,427,939 | 101 | 44 | 43,505,608 | 36–101 | 44 | 1.97 | 80.21 | 96.17 | |
| F1.T1.3.4.A | 39,689,690 | 101 | 44 | 38,969,075 | 36–101 | 44 | 1.82 | 84.36 | 96.60 | |
| F1.T1.6.7.B | 39,060,576 | 101 | 44 | 38,311,420 | 36–101 | 43 | 2.19 | 86.31 | 96.84 | |
| F1.T1.9.7.A | 41,154,600 | 101 | 44 | 40,401,529 | 36–101 | 44 | 1.82 | 86.89 | 96.84 | |
| F2.T1.10.4.A | 39,239,798 | 101 | 44 | 38,498,417 | 36–101 | 43 | 1.89 | 88.51 | 97.01 | |
| F2.T1.11.0.B | 40,638,650 | 101 | 44 | 39,844,781 | 36–101 | 44 | 1.95 | 88.57 | 97.02 | |
| F2.T1.3.4.B | 40,749,175 | 101 | 44 | 39,928,436 | 36–101 | 44 | 2.01 | 88.26 | 96.85 | |
| F2.T1.6.7.B | 41,118,657 | 101 | 44 | 40,283,656 | 36–101 | 44 | 2.03 | 88.99 | 96.99 | |
| F2.T1.9.7.A | 37,606,367 | 101 | 44 | 36,768,140 | 36–101 | 45 | 2.23 | 89.85 | 97.19 | |
| Treat | F1.T4.10.5.B | 36,940,490 | 101 | 44 | 36,167,170 | 36–101 | 44 | 2.10 | 90.05 | 96.70 |
| F1.T4.11.2.A | 39,943,939 | 101 | 44 | 39,102,409 | 36–101 | 44 | 2.08 | 88.52 | 96.84 | |
| F1.T4.10.4.B | 42,127,626 | 101 | 44 | 41,278,975 | 36–101 | 44 | 2.01 | 87.22 | 96.60 | |
| F1.T4.16.4.A | 37,505,867 | 101 | 44 | 36,701,336 | 36–101 | 43 | 2.15 | 88.47 | 96.75 | |
| F1.T4.23.3.B | 40,108,579 | 101 | 44 | 39,263,353 | 36–101 | 44 | 2.11 | 87.73 | 96.55 | |
| F2.T4.10.5.B | 38,137,968 | 101 | 45 | 37,295,092 | 36–101 | 45 | 2.21 | 88.03 | 96.79 | |
| F2.T4.10.2.B | 37,911,150 | 101 | 44 | 37,094,121 | 36–101 | 44 | 2.16 | 88.99 | 96.87 | |
| F2.T4.16.4.A | 38,295,985 | 101 | 44 | 37,450,553 | 36–101 | 44 | 2.21 | 89.22 | 96.97 | |
| F2.T4.19.A | 38,094,651 | 101 | 44 | 37,303,953 | 36–101 | 43 | 2.08 | 88.57 | 97.00 | |
| F2.T4.23.3.B | 39,509,036 | 101 | 46 | 38,845,012 | 36–101 | 44 | 1.96 | 90.58 | 97.09 | |
| Average | 39,286,885 | 101 | 44.5 | 38,530,418 | 36–101 | 44.4 | 1.93 | 87.62 | 96.78 | |
GC, guanine-cytosine
생물학적 과정(BP, Biological Process)에서 유의한 GO term들을 Fig 2에서 확인하였고, 각 term들의 지표를 Table 3과 4에 나타내었다. 주 차별 GO Treemap을 분석할 때 고온자극여부에 초점을 맞춰 연관 분석을 수행하였다. 1주 차의 Treemap에서는 TLR9이 관여하는 “toll-like receptor 9 signaling pathway”이 treemap의 크기가 크고, Count 값이 많은 것으로 보아 1주 차에서 가장 유의한 것으로 확인된다 (Fig 2A, Table 3). 2주 차에서 Count 값이 가장 많은 BP인 “regulation of cell cycle”은 호르몬을 조절하여 긍정적 혹은 부정적 역할을 하는 것으로 보이며, 그 외에 “Ammonium transmembrane transport”, “heme biosynthetic process”가 P-value 값이 낮고, treemap의 크기가 커 유의할 것으로 나타났다 (Fig 2B, Table 4).
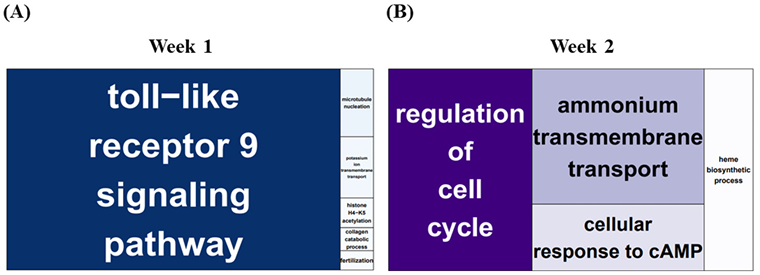
Figure 2. Gene Ontology biological process treemap of pig samples in week 1 (A) and week 2 (B).
Table 3. Index of the gene ontology biological process term in week 1.
| Term | Count | % | P-value | Genes | Fold Enrichment |
|---|---|---|---|---|---|
| GO:0034162 toll-like receptor 9 signaling pathway |
14 | 5.88 | 1.68E-21 | TLR9 | 59.38 |
FDR, False Discovery Rate
Table 4. Index of the gene ontology biological process term in week 2.
| Term | Count | % | P-value | Genes | Fold Enrichment |
|---|---|---|---|---|---|
| GO:0072488 ammonium transmembrane transport | 3 | 4.41 | 1.38E-4 | RHAG, RHCE, AQP1 | 156.72 |
| GO:0006783 heme biosynthetic process | 3 | 4.41 | 8.95E-4 | ATP5IF1, ALAS2, FECH | 65.30 |
| GO:0071320 cellular response to cAMP | 3 | 4.41 | 5.29E-3 | PDE2A, PKD2, AQP1 | 27.02 |
| GO:0051726 regulation of cell cycle | 4 | 5.88 | 0.02 | INO80C, FOXM1, CCNDBP1, MASTL | 7.57 |
| GO:0015670 carbon dioxide transport | 2 | 2.94 | 0.02 | RHAG, AQP1 | 104.48 |
| GO:0009992 cellular water homeostasis | 2 | 2.94 | 0.02 | MIOX, AQP1 | 87.06 |
| GO:0060315 negative regulation of ryanodine-sensitive calcium-release channel activity | 2 | 2.94 | 0.02 | PKD2, CLIC2 | 87.06 |
| GO:0071805 potassium ion transmembrane transport | 3 | 4.41 | 0.03 | PKD2, KCNH1, AQP1 | 11.87 |
| GO:0010923 negative regulation of phosphatase activity | 2 | 2.94 | 0.03 | TMEM225B | 65.30 |
| GO:0019934 cGMP-mediated signaling | 2 | 2.94 | 0.05 | PDE2A, AQP1 | 34.83 |
| GO:0032147 activation of protein kinase activity | 2 | 2.94 | 0.07 | TPX2, STRADB | 24.98 |
| GO:2000134 negative regulation of G1/S transition of mitotic cell cycle | 2 | 2.94 | 0.08 | PKD2, EZH2 | 24.88 |
| GO:0007094 mitotic spindle assembly checkpoint | 2 | 2.94 | 0.08 | BUB1B, TTK | 23.74 |
FDR, False Discovery Rate
유의한 pathway는 KEGG database를 기반으로 확인하였다 (Fig 3). 주 차별 돼지 샘플 간의 유사성과 특성에 대한 연관성 분석을 실시하였다. 1주 차 KEGG pathway들을 봤을 때, 대부분의 pathway들이 암과 관련된 pathway를 나타내며, “Focal adhesion”을 제외한 나머지 pathway들에 APC2와 TCFL2가 관여하는 것을 알 수 있었다 (Fig 3A, Table 5). 2주 차 pathway에는 “Metabolic pathways”만 존재하며, 이에 관여하는 유전자들로는 MIOX, CA1, ALAS2, GPX1, FECH, ARG1, PDE2A, MTMR8, EZH2들이 있다 (Fig 3A, Table 6).
BP에 대한 주차 별 DEG들의 공동 발현 네트워크를 Cluego와 CluePedia를 통해 시각화하였다 (Fig 4). Node의 크기는 DEG들이 많이 관여하는 BP일수록 크며, 이는 node의 크기가 클수록 유의하다는 것을 나타낸다. Week 1을 보면, NOTCH1, ENG, SMAD7들은 DEG들 중 가장 많은 BP에 관여한다 (Fig 4).
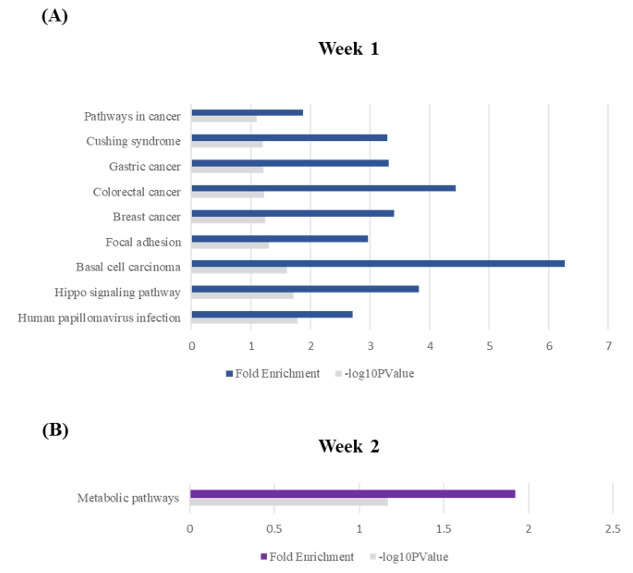
Figure 3. KEGG pathways of pig samples in week 1 (A) and week 2 (B).
Table 5. Index of the KEGG pathways in week 1.
| Term | Count | % | P-value | Genes | Fold Enrichment |
|---|---|---|---|---|---|
| ssc05165 Human papillomavirus infection |
9 | 3.78 | 0.02 | APC2, TCF7L2, NOTCH1, FZD4, COL4A4, SPP1, PPP2R5D, SCRIB, AXIN2 | 2.71 |
| ssc04390 Hippo signaling pathway |
6 | 2.52 | 0.02 | APC2, TCF7L2, FZD4, SCRIB, AXIN2, SMAD7 | 3.82 |
| ssc05217 Basal cell carcinoma |
4 | 1.68 | 0.03 | APC2, TCF7L2, FZD4, AXIN2 | 6.27 |
| ssc04510 Focal adhesion |
6 | 2.52 | 0.05 | FLT1, COL4A4, SPP1, RAPGEF1, ZYX, MYL9 | 2.96 |
| ssc05224 Breast cancer |
5 | 2.10 | 0.06 | APC2, TCF7L2, NOTCH1, FZD4, AXIN2 | 3.40 |
| ssc05210 Colorectal cancer |
4 | 1.68 | 0.06 | APC2, TCF7L2, AXIN2, RALGDS | 4.44 |
| ssc05226 Gastric cancer |
5 | 2.10 | 0.06 | APC2, TCF7L2, JUP, FZD4, AXIN2 | 3.31 |
| ssc04934 Cushing syndrome |
5 | 2.10 | 0.07 | APC2, TCF7L2, FZD4, ADCY4, AXIN2 | 3.29 |
| ssc05200 Pathways in cancer |
10 | 4.20 | 0.08 | APC2, TCF7L2, NOTCH1, JUP, FZD4, COL4A4, ADCY4, ABL1, AXIN2, RALGDS | 1.87 |
FDR, False Discovery Rate
Table 6. Index of the KEGG pathways in week 2.
| Term | Count | % | P-value | Genes | Fold Enrichment |
|---|---|---|---|---|---|
| ssc01100 Metabolic pathways |
9 | 13.24 | 0.067 | MIOX, CA1, ALAS2, GPX1, FECH, ARG1, PDE2A, MTMR8, EZH2 | 1.92 |
FDR, False Discovery Rate
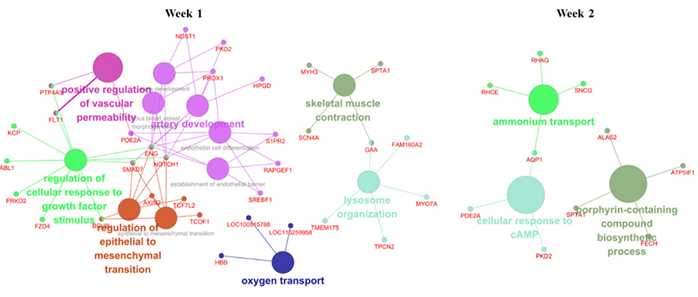
Figure 4. Cluego network analysis of 2 weeks of pig samples.
고온 스트레스는 다양한 측면에서 돼지에게 해로운 영향을 미치고 있으며, 지구 온난화의 가속화에 따라 고온 스트레스로 인한 돼지의 질병 및 건강에 대한 고려가 필요하다. 본 연구에서는 고온 스트레스에 노출된 돼지의 생리적 과정을 이해하기 위해 샘플을 대조군과 실험군으로 구분하여 주차별 시료의 유전자 발현 수준의 차이를 조사하였다. 이를 통해, 고온 스트레스에 관한 생물학적 pathway를 해석하여 포괄적인 단서를 제공한다.
표현형 분석 결과를 기반으로 고온스트레스가 돼지의 생산성에 미치는 영향을 확인했을 때, 평균 일당 증체량 및 평균 일당 사료 섭취량은 대조군에서 유의하게 높았다. 고온 스트레스의 영향으로 비육돈의 사료 섭취량이 감소하였고, 이에 따라 평균 일일 증체량도 감소한 것으로 알 수 있다 (Table 1). DEG profiling 결과로 1주 차에는 354개의 DEG, 2주 차에는 99개의 DEG들이 있으며, 1주 차에는 상향 조절된 DEG가 하향 조절된 DEG보다 많고, 2주 차에는 하향 조절된 DEG가 상향 조절된 DEG보다 많다 (Fig 1). 이를 통해, 1주 차에서는 고온 스트레스에 연관된 유의미한 유전자가 풍부하게 존재한다는 것을 알 수 있다. 1주 차 treemap에서 “tolllike receptor 9 signaling pathway”가 가장 유의하고, 이 pathway에 관여하는 유전자가 TLR9임을 알 수 있다 (Fig 2A, Table 3). TLR9 발현은 고온 스트레스가 발생할 때 증가하며 고온으로 인한 세포 손상으로부터 신체를 방어하는 데 중요한 역할을 한다 (Chen et al., 2005). 이를 통해, 1주 차에서 고온 스트레스에 대한 돼지의 면역반응이 TLR9의 메커니즘에 기초하여 발생하였다는 것을 알 수 있었다. 1주 차의 KEGG pathway들에는 다양한 pathway들이 나타나며, 이 pathway들 중 “Focal adhesion”을 제외한 나머지 pathway 모두 APC2 및 TCF7L2와 관련이 있다 (Fig 3A, Table 5). APC2는 고온 스트레스 환경에서 Wnt signalling pathway를 조절한다 (Livernois et al., 2021). TCF7L2는 Wnt signalling pathway와 관련되어 있으며, 이는 지질 및 포도당 대사에 관여하는 유전자 발현을 조절한다 (Del Bosque-Plata et al., 2021). 고온 스트레스는 일반적으로 돼지의 에너지 대사에 변화를 일으키며, 이 과정에서 에너지가 지방으로 축적되어 돼지의 대사율 감소 및 지질 합성 활성화로 이어진다 (Qu et al. 2015). 특히, TCF7L2는 지질 합성 경로의 조절에 관여하여 돼지의 비정상적인 지방 생성을 유발하여 경제적 가치하락으로 이어진다 (Du et al., 2009). 고온 스트레스 환경에서의 에너지 축적이 돼지의 비가식 지방 증가와 높은 연관성을 가진다는 사실은 선행 연구에서 보고된 바 있으며, 본 연구에서도 동일한 경향을 확인하였다. 이러한 결과는 고온 스트레스가 경제적 형질에 미치는 부정적 영향을 완화하기 위해 적정 온도를 유지하거나 스트레스를 줄일 수 있는 환경을 조성하는 것의 중요성을 시사한다. 따라서, 사육 환경의 적절한 조절은 고온 스트레스로 인한 생산성 저하를 방지하는 주요 요소로 고려될 수 있음을 본 연구에서 제시하였다.
ALAS2와 FECH는 2주 차 bp의 “heme biosynthetic process”, KEGG pathway의 “Metabolic pathways”에 모두 관여하는 유전자로 (Table 4, 6), heme biosynthetic에서 산소 결합 및 운반 역할을 하는 heme 합성에 영향을 미친다 (Dutt et al., 2022). 선행연구에서는 고온 스트레스를 받은 돼지에서 적혈구 손상 증가 및 사료 섭취 감소로 인해 헤모글로빈농도가 감소할 수 있다고 보고되었는데 (Cui et al., 2019), 실제 DEG profiling 결과 ALAS2와 FECH는 하향 조절되어 Control 그룹에서 더 발현되었다 (Table 7). Cluego 네트워크 분석으로 여러 BP와 동시에 네트워크를 이루는 유의한 DEG를 확인할 수 있다. 유의한 DEG 중 하나인 NOTCH1의 활성화는 TGF-β signaling을 촉진 및 증가시키며 (Asano et al., 2008), TGF-β signaling은 주로 면역억제역할을 한다 (Johnston et al., 2016). 이를 통해, 고온 스트레스로 인해 돼지 내에서 면역 반응이 증가하여 이에 대한 반응으로 TGF-β signaling가 증가해 면역억제 과정이 이루어졌다고 해석할 수 있다. 선행연구에서는 시간이 지날수록 돼지 고온 스트레스에 대한 반응으로 체내 구성을 변화시킨다고 보고되었다 (Ross et al., 2015). 따라서 본 연구에서, 1주 차에서 TLR9, NOTCH1 등 DEG들의 발현을 미루어 보아, 고온 스트레스가 미치는 부정적 영향을 확인하였다. 결론적으로, 비육돈의 고온 스트레스에 대한 DEG profiling 및 기능 분석 결과로 고온 스트레스에 특이적인 후보 유전자 바이오마커를 발굴할 수 있었으며, 다양한 BP 및 생물학적 pathway에 영향을 미치는 것을 알 수 있었다.
Table 7. Index of “ALAS2”, “FECH’ gene in week 2 DEG.
| Gene_Symbol | Gene_name | logFC | P-value | FDR |
|---|---|---|---|---|
| ENSSSCG00000004541 | FECH | -1.23 | 6.17E-06 | 1.67E-3 |
| ENSSSCG00000012347 | ALAS2 | -1.32 | 4.57E-07 | 1.90E-4 |
FDR, False Discovery Rate
본 연구에서 우리는 고온 스트레스 영향을 받은 돼지의 RNA-Seq 및 기능 분석을 수행하였다. 돼지 전혈의 전사체를 분석하여 DEG를 검출하고, 이를 이용하여 pathway, 생물학적 과정 등을 시각화하였고, 시각화한 분석 결과들을 토대로 DEG들의 생물학적 기능을 예측해 보았다. 그 결과, 1주 차에서 “toll-like receptor 9 signaling”에 관여하며 열로 인한 세포 손상으로부터 신체를 방어하는 TLR9를 확인하였고, Wnt signalling pathway에 연관되어 있는 APC2, TCF7L2를 식별하였다. 또한, Cluego를 통해, NOTCH1을 확인하고, 고온 스트레스로 인한 면역반응에 대한 면역억제작용이 발생하였다는 시선을 제시한다. 2주 차에서는 산소 결합 및 운반 역할을 하는 heme 합성에 영향을 미치는 ALAS2와 FECH를 확인하였다. 본 연구를 통해 돼지의 고온 스트레스 메커니즘에서 발생하는 생물학적 과정에 관여하는 후보유전자를 제시하여 고온 스트레스 해결책에 대한 포괄적인 이해를 제시하는 것에 의의가 있다.
No potential conflict of interest relevant to this article is reported.
This work was carried out with the support of the Cooperative Research Program for Agriculture Science & Technology Development, of the Rural Development Administration, Republic of Korea (PJ01620403).
Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data: Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom.
Asano N, Watanabe T, Kitani A, Fuss IJ, Strober W. 2008. Notch1 Signaling and Regulatory T Cell Function. The Journal of Immunology 180:27962804.
[DOI][PubMed]
Bindea G, Galon J, Mlecnik B. 2013. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 29:661-663.
[DOI][PubMed][PMC]
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, Galon J. 2009. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091-1093.
[DOI][PubMed][PMC]
Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114-2120.
[DOI][PubMed][PMC]
Cantet JM, Yu Z, Ríus AG. 2021. Heat Stress-Mediated Activation of Immune-Inflammatory Pathways. Antibiotics 10:1285.
[DOI][PubMed][PMC]
Chen W, Wang J, An H, Zhou J, Zhang L, Cao X. 2005. Heat shock up-regulates TLR9 expression in human B cells through activation of ERK and NF-κB signal pathways. Immunology letters 98:153-159.
[DOI][PubMed]
Consortium GO. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic acids research 32:D258-D261.
[DOI][PubMed][PMC]
Cui Y, Wang C, Hao Y, Gu X, Wang H. 2019. Chronic Heat Stress Induces Acute Phase Responses and Serum Metabolome Changes in Finishing Pigs. Animals 9:395.
[DOI][PubMed][PMC]
Del Bosque-Plata L, Martínez-Martínez E, Espinoza-Camacho M, Gragnoli C. 2021. The Role of TCF7L2 in Type 2 Diabetes. Diabetes 70:12201228.
[DOI][PubMed][PMC]
Du Z-Q, Fan B, Zhao X, Amoako R, Rothschild MF. 2009. Association Analyses Between Type 2 Diabetes Genes and Obesity Traits in Pigs. Obesity 17:323-329.
[DOI][PubMed]
Dutt S, Hamza I, Bartnikas TB. 2022. Molecular Mechanisms of Iron and Heme Metabolism. Annual Review of Nutrition 42:311-335.
[DOI][PubMed][PMC]
Godyń D, Herbut P, Angrecka S, Corrêa Vieira FM. 2020. Use of Different Cooling Methods in Pig Facilities to Alleviate the Effects of Heat Stress-A Review. Animals 10:1459.
[DOI][PubMed][PMC]
Harrison PW, Wright AE, Mank JE. 2012. The evolution of gene expression and the transcriptome-phenotype relationship. Seminars in Cell & Developmental Biology 23:222-229.
[DOI][PubMed][PMC]
Hrdlickova R, Toloue M, Tian B. 2017. RNA‐Seq methods for transcriptome analysis. Wiley Interdisciplinary Reviews: RNA 8:e1364.
[DOI][PubMed][PMC]
Huo C, Xiao C, She R, Liu T, Tian J, Dong H, Tian H, Hu Y. 2019. Chronic heat stress negatively affects the immune functions of both spleens and intestinal mucosal system in pigs through the inhibition of apoptosis. Microbial pathogenesis 136:103672.
[DOI][PubMed]
Ingram D. 1965. Evaporative cooling in the pig. Nature 207:415-416.
[DOI][PubMed]
Johnston CJC, Smyth DJ, Dresser DW, Maizels RM. 2016. TGF-β in tolerance, development and regulation of immunity. Cellular Immunology 299:14-22.
[DOI][PubMed][PMC]
Jung HS, Choi Y, Oh JH, Lim GH. 2002. Recent trends in temperature and precipitation over South Korea. 22:1327-1337.
[DOI]
KERI (KOREA RURAL ECONOMIC INSTITUTE). 2023. 2023. 06 Pig Industry Observation Information. KERI, Naju, Korea [in Korean]
Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28:27-30.
[DOI][PubMed][PMC]
Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12:357-360.
[DOI][PubMed][PMC]
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078-2079.
[DOI][PubMed][PMC]
Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923-930.
[DOI][PubMed]
Livernois AM, Mallard BA, Cartwright SL, Cánovas A. 2021. Heat stress and immune response phenotype affect DNA methylation in blood mononuclear cells from Holstein dairy cows. Scientific Reports 11:11371.
[DOI][PubMed][PMC]
Luo H, Li X, Hu L, Xu W, Chu Q, Liu A, Guo G, Liu L, Brito LF, Wang Y. 2021. Genomic analyses and biological validation of candidate genes for rectal temperature as an indicator of heat stress in Holstein cattle. Journal of Dairy Science 104:4441-4451.
[DOI][PubMed]
Marguerat S, Bähler J. 2010. RNA-Seq: from technology to biology. Cellular and Molecular Life Sciences 67:569-579.
[DOI][PubMed][PMC]
Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M. 2015. The human transcriptome across tissues and individuals. Science 348:660-665.
[DOI][PubMed][PMC]
Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLOS ONE 7:e30619.
[DOI][PubMed][PMC]
Pearce SC, Mani V, Boddicker RL, Johnson JS, Weber TE, Ross JW, Rhoads RP, Baumgard LH, Gabler NK. 2013. Heat Stress Reduces Intestinal Barrier Integrity and Favors Intestinal Glucose Transport in Growing Pigs. PLOS ONE 8:e70215.
[DOI][PubMed][PMC]
Perini F, Cendron F, Rovelli G, Castellini C, Cassandro M, Lasagna E. 2021. Emerging Genetic Tools to Investigate Molecular Pathways Related to Heat Stress in Chickens: A Review. Animals 11:46.
[DOI][PubMed][PMC]
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-Sequencing and microarray studies. Nucleic Acids Research 43:e47-e47.
[DOI][PubMed][PMC]
Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139-140.
[DOI][PubMed][PMC]
Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-Seq data. Genome Biology 11:1-9.
[DOI][PubMed][PMC]
Ross JW, Hale BJ, Gabler NK, Rhoads RP, Keating AF, Baumgard LH. 2015. Physiological consequences of heat stress in pigs. Animal Production Science 55:1381-1390.
[DOI]
SAS Institute Inc. 2016. SAS® 9.4 Language Reference: Concepts, Sixth Edition. SAS Institute Inc., Cary, NC, USA.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498-2504.
[DOI][PubMed][PMC]
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W. 2022. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res 50:W216-W221.
[DOI][PubMed][PMC]
Wickham H. 2011. ggplot2. Wiley Interdisciplinary Reviews: Computational Statistics 3:180-185.
[DOI]
Witte D, Ellis M, McKeith F, Wilson EJJoAS. 2000. Effect of dietary lysine level and environmental temperature during the finishing phase on the intramuscular fat content of pork. Journal of Animal Science 78:1272-1276.
[DOI][PubMed]
Wolp RC, Rodrigues NEB, Zangeronimo MG, Cantarelli VS, Fialho ET, Philomeno R, Alvarenga RR, Rocha LF. 2012. Soybean oil and crude protein levels for growing pigs kept under heat stress conditions. Livestock Science 147:148-153.
[DOI]
Xin H, Harmon J. 1998. Livestock industry facilities and environment: heat stress indices for livestock.